Neuromuscular disorders associated with monoclonal gammopathies are usually uncovered in approximately 10% of patients presenting with peripheral neuropathy complaints. This discovery should prompt further evaluation for underlying plasma cell dyscrasias. The most frequent monoclonal disorders associated with neuropathy are smoldering myeloma, multiple myeloma, Waldenström macroglobulinemia, solitary plasmacytoma, systemic immunoglobulin light chain (AL) amyloidosis, POEMS (polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes), and cryoglobulinemia. If these are excluded by careful evaluation the patient is classified as having monoclonal gammopathy of undetermined significance. Diagnostic criteria, risk stratification to determine prognosis, and current management for these disorders are reviewed in this article.
The broad spectrum of plasma cell dyscrasias characterized as paraproteinemias or monoclonal gammopathies is subclassified by proliferation of particular individual clones of β lymphocytes that secrete excess monoclonal antibodies. This overproduction of plasma cells can be either neoplastic or a low-grade potentially neoplastic process. Monoclonal proteins are prevalent in 3% of the general population older than 50 years of age, with rates increasing with age. Prevalence rates are higher for men than women, and African Americans have a threefold higher age-adjusted prevalence rate than Caucasian persons .
Paraproteinemias are usually asymptomatic and are found on routine blood testing without associated abnormalities. Nevertheless, certain disease associations occur more often than by chance and include: (1) monoclonal gammopathies of undetermined significance (MGUS), biclonal gammopathies, idiopathic Bence Jones proteinuria; (2) the malignant monoclonal gammopathies of multiple myeloma, smoldering multiple myeloma, Waldenström macroglobulinemia, uncommon malignant disorders of solitary plasmacytoma, and POEMS syndrome ( p olyneuropathy, o rganomegaly, e ndocrinopathy, m onoclonal gammopathy, and s kin changes); (3) systemic AL (immunoglobulin light chain) amyloidosis; (4) cryoglobulinemia; and (5) heavy-chain diseases . When clinical features are present, they range from fatigue, weight loss, purpura, congestive heart failure, nephrotic syndrome, peripheral neuropathy, and orthostatic hypotension, to mucocutaneous bleeding .
Paraprotein-associated neuropathies have emerged as an important category of late-onset chronic polyneuropathies, warranting further evaluation for an underlying plasma cell dyscrasia . In patients presenting with peripheral neuropathy of unknown cause, 10% will have a monoclonal protein, the majority of whom will be found to have MGUS. This is of even greater importance in patients with patterns of nerve conduction abnormality suggestive of demyelination, autonomic dysfunction, predominantly motor neuropathy, or lower motor neuron findings. The plasma cells disorders most often associated with neuropathy include MGUS, smoldering myeloma, multiple myeloma, Waldenström macroglobulinemia, solitary plasmacytoma, systemic AL amyloidosis and POEMS and are discussed later in this monograph .
It is helpful to distinguish between a monoclonal and a polyclonal process in plasma cell disorders, because unlike the monoclonal process, a reactive or inflammatory process commonly causes the polyclonal increase in immunoglobulin (Ig) . Polyclonal immunoglobulins are created by many clones of plasma cells. Blood levels of polyclonal gamma globulin of 3 g/dL or more are associated with liver disease, connective tissue diseases, chronic infections, and nonhematologic malignancies .
The monoclonal process, however, is more often associated with premalignant or malignant disease than its polyclonal counterpart. There can be an overlap in antibody-mediated pathogenic mechanisms among these different monoclonal and polyclonal antibodies .
Monoclonal proteins (also called M proteins, myeloma proteins, or paraproteins) consist of two heavy polypeptide chains of the same class and subclass and two light polypeptide chains of the same type. The different monoclonal proteins are known by letters that correspond to the class of their heavy chains, which are designated by Greek alphabet characters: γ (gamma) in IgG, α (alpha) in IgA, μ (mu) in IgM, δ (delta) in immunoglobulin D (IgD), and ϵ (epsilon) in immunoglobulin E (IgE). There are four subclasses for IgG, two subclasses for IgA, and no subclasses for IgM, IgD, or IgE. The light-chain types are kappa (κ) and lambda (λ). Each of these monoclonal proteins is produced by a proliferation of single clonal population of plasma cells in the bone marrow. The mechanism of monoclonal expansion of a single Ig-secreting plasma cell population is unknown in most instances . A review of the data by Kyle and Rajkumar , however, suggests in at least 50% of MGUS there is evidence of genomic instability on molecular genetic testing, including primary chromosomal translocations at the immunoglobulin heavy chain locus 14q32 (50%), hyperdiploidy (40%), or unknown (10%) . The cause for this genomic instability is uncertain. Current evidence, however, suggests that antigenic stimulation related to infection and immune dysregulation may be an important factor . Additional contributors to the progression of MGUS to multiple myeloma are under study and include changes in the microenvironment by way of induction of angiogenesis, suppression of cell-mediated immunity, adhesions of myeloma cells to stroma, alteration of adhesion molecules, and stromal cytokine overexpression .
Increased osteoclast activation and receptor activator of nuclear factor-kappa beta B ligand (RANKL) and decreased levels of osteoprotegerin (OPG) result in lytic bone lesions and osteoporosis . The pathogenic link between the monoclonal proteins and nerve damage is known only in a few instances. In the IgM class it is believed that neuropathy is related to the reactivity of the circulating antibodies that are directed against specific neural antigens expressed on the peripheral nerves, such as myelin-associated glycoprotein (MAG), chondroitin sulfate, and sulfatide with consequential complement-dependent nerve damage . Findings have shown that the deposition of IgM protein within the myelin in large and small myelinated fibers may have a role in the pathogenesis of neuropathy, although this remains unproven .
Recognition of monoclonal proteins
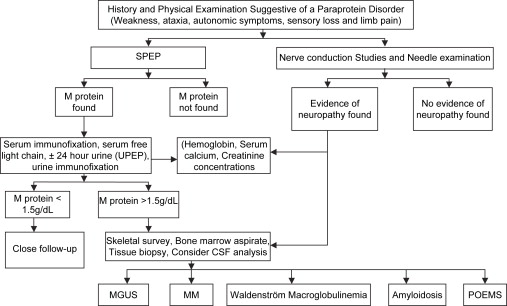
Identification of M proteins is best achieved by serum protein electrophoresis techniques. The immunofixation process distinguishes the immunoglobulin class and type of light chain. Densitometry is used to measure the monoclonal protein that is visible on serum electrophoresis and has replaced the use of nephelometry as a more reliable way to measure immunoglobulin levels. A 24-hour analysis of urine for protein excretion and a urine protein electrophoresis and immunofixation is warranted to detect and quantify the monoclonal protein in the urine. To date this has been the standard recommendation . A recent study by Katzmann and colleagues suggests this may not be necessary because of the highly sensitive serum free light-chain assay now available. Measurement of β 2 -microglobulin has not proved predictive of malignant transformation and is no longer recommended. If a monoclonal protein is found, additional hematologic studies (serum calcium, complete blood count, and serum creatinine) are needed. Although a skeletal survey and aspirated bone marrow biopsy are generally performed to rule out myeloma, they are not considered necessary when other laboratory tests are normal and the serum monoclonal spike is less than 1.5 g per deciliter and other laboratory tests are normal . If abnormalities are detected on these tests, appropriate tissue biopsy is recommended to exclude amyloidosis. When no monoclonal protein is uncovered, patients who have motor polyradiculoneuropathy with demyelinating features on nerve conduction studies should have cerebrospinal fluid examination, skeletal bone survey, and sural nerve biopsy ( Fig. 1 ). Sural nerve biopsy has shown mixed pathology (fiber loss, segmental demyelination, and axonal degeneration). In the IgM class a predominantly demyelinating process is often observed. Patients who have progressive axonal neuropathies with autonomic dysfunction require biopsy of one or more appropriate tissues to rule out or confirm amyloidosis .
Electromyographic studies
Electromyographic changes in plasma cell disorders show abnormalities consistent with demyelination and axonal degeneration ( Table 1 ). Nerve conductions are typically abnormal in motor and sensory fibers in the upper and lower extremities. Motor nerve conduction velocities are often decreased to less than the lower limits of normal by 20% or more. Sensory nerve action potentials are consistently reduced in amplitude or are unobtainable, with more pronounced changes in the lower limb nerves. Frequently F-wave latencies are prolonged but not out of proportion to the degree of peripheral nerve conduction slowing. In IgM neuropathies, the nerve conduction abnormalities tend to be more severe than the cases involving other protein classes. Needle examination demonstrates changes consistent with active denervation (increased insertional activity with fibrillation potentials) in more than 80% of patients. Evidence of coexisting demyelination and denervation is common. The sural sensory nerve more often shows damage than the median sensory nerve .
| Disorder | Anatomical pattern | Nerve conduction changes | Pathological features | Paraprotein | Clinical features | Treatment |
|---|---|---|---|---|---|---|
| MGUS | DS, DSSP | Axonal | Axonal degeneration | IgM-κ or IgG-κ | Asymptomatic, neuropathy | Surveillance |
| Smoldering multiple myeloma | DS, DSSP | Axonal | Axonal degeneration, with or without amyloid deposition | IgM-κ or IgG-κ | Asymptomatic, neuropathy | Surveillance |
| Multiple myeloma | DS, DSSP | Axonal | Axonal degeneration, with or without amyloid deposition | IgM-κ or IgG-κ | Bone pain, fatigue, anemia, polyneuropathy, hypercalcemia, renal insufficiency | Irradiation, chemotherapy, autologous stem cell transplantation and novel drug trials (Thalidomide) |
| Solitary plasmacytoma | DS, DSSP | Axonal | Axonal degeneration, with or without amyloid deposition | Polyneuropathy, bone pain, solitary bone lesion | Resection and irradiation | |
| POEMS syndrome | DS, DSSP areflexia simulates CIDP | Axonal, Demyelinating, similar to CIDP | Axonal, sclerotic bone lesions | IgG- λ or IgA-λ | Polyneuropathy, organomegaly, endocrinopathy, skin changes, peripheral edema, fatigue, papilledema, lymph node hyperplasia | Irradiation, autologous stem cell transplantation, melphalan plus prednisone |
| Waldenström macroglobulinemia | DS, DSSP a simulates CIDP | Axonal, demyelinating | IgM-κ | Fatigue, weight loss, oronasal bleeding, visual blurring, dyspnea, polyneuropathy, encephalopathy | Rituximab, nucleoside analogues, alkator agents | |
| Systemic AL amyloidosis | S, A | Axonal | Axonal degeneration with amyloid deposition, rare myopathy | IgG-λ or IgA-λ | Chronic painful polyneuropathies, systemic organ involvement, rare proximal muscle weakness | Melphalan plus prednisone |
| MMN | S, SM | M | Lower motor neuron | IgM-GM1 | Limb weakness, wasting predominantly in the arms | IVIg |
| Cryoglobulinemia | S, M, DSSP, SM, MM | Axonal | Axonal degeneration, vasculitis, inflammatory infiltrate | IgM or IgG | Arthralgias, purpura, leg ulcers, Raynaud’s phenomenon, hepatosplenomegaly, painful polyneuropathies | For mild conservative measures. For severe plasma exchange, Prednisone, cytotoxic therapy |
Electromyographic studies
Electromyographic changes in plasma cell disorders show abnormalities consistent with demyelination and axonal degeneration ( Table 1 ). Nerve conductions are typically abnormal in motor and sensory fibers in the upper and lower extremities. Motor nerve conduction velocities are often decreased to less than the lower limits of normal by 20% or more. Sensory nerve action potentials are consistently reduced in amplitude or are unobtainable, with more pronounced changes in the lower limb nerves. Frequently F-wave latencies are prolonged but not out of proportion to the degree of peripheral nerve conduction slowing. In IgM neuropathies, the nerve conduction abnormalities tend to be more severe than the cases involving other protein classes. Needle examination demonstrates changes consistent with active denervation (increased insertional activity with fibrillation potentials) in more than 80% of patients. Evidence of coexisting demyelination and denervation is common. The sural sensory nerve more often shows damage than the median sensory nerve .
| Disorder | Anatomical pattern | Nerve conduction changes | Pathological features | Paraprotein | Clinical features | Treatment |
|---|---|---|---|---|---|---|
| MGUS | DS, DSSP | Axonal | Axonal degeneration | IgM-κ or IgG-κ | Asymptomatic, neuropathy | Surveillance |
| Smoldering multiple myeloma | DS, DSSP | Axonal | Axonal degeneration, with or without amyloid deposition | IgM-κ or IgG-κ | Asymptomatic, neuropathy | Surveillance |
| Multiple myeloma | DS, DSSP | Axonal | Axonal degeneration, with or without amyloid deposition | IgM-κ or IgG-κ | Bone pain, fatigue, anemia, polyneuropathy, hypercalcemia, renal insufficiency | Irradiation, chemotherapy, autologous stem cell transplantation and novel drug trials (Thalidomide) |
| Solitary plasmacytoma | DS, DSSP | Axonal | Axonal degeneration, with or without amyloid deposition | Polyneuropathy, bone pain, solitary bone lesion | Resection and irradiation | |
| POEMS syndrome | DS, DSSP areflexia simulates CIDP | Axonal, Demyelinating, similar to CIDP | Axonal, sclerotic bone lesions | IgG- λ or IgA-λ | Polyneuropathy, organomegaly, endocrinopathy, skin changes, peripheral edema, fatigue, papilledema, lymph node hyperplasia | Irradiation, autologous stem cell transplantation, melphalan plus prednisone |
| Waldenström macroglobulinemia | DS, DSSP a simulates CIDP | Axonal, demyelinating | IgM-κ | Fatigue, weight loss, oronasal bleeding, visual blurring, dyspnea, polyneuropathy, encephalopathy | Rituximab, nucleoside analogues, alkator agents | |
| Systemic AL amyloidosis | S, A | Axonal | Axonal degeneration with amyloid deposition, rare myopathy | IgG-λ or IgA-λ | Chronic painful polyneuropathies, systemic organ involvement, rare proximal muscle weakness | Melphalan plus prednisone |
| MMN | S, SM | M | Lower motor neuron | IgM-GM1 | Limb weakness, wasting predominantly in the arms | IVIg |
| Cryoglobulinemia | S, M, DSSP, SM, MM | Axonal | Axonal degeneration, vasculitis, inflammatory infiltrate | IgM or IgG | Arthralgias, purpura, leg ulcers, Raynaud’s phenomenon, hepatosplenomegaly, painful polyneuropathies | For mild conservative measures. For severe plasma exchange, Prednisone, cytotoxic therapy |
Monoclonal gammopathy of undetermined significance
MGUS is the most common plasma cell dyscrasia found in the general population and usually presents asymptomatically after the fifth decade of life . It is considered a premalignant disorder characterized by limited monoclonal plasma cell proliferation in the bone marrow and absence of end-organ damage. In de novo paraproteinemia, approximately two thirds of the time after exclusion of amyloidosis, multiple or osteosclerotic myeloma, Waldenström macroglobulinemia, lymphoma, or lymphoproliferative disease, no identifiable cause is found, and the disorder by exclusion is classified as MGUS. Although characterized as premalignant, it is not truly benign in that it is linked to a lifelong risk for progression to multiple myeloma or related disorders, making lifelong follow-up necessary in all persons .
An M protein is found in 3% to 4% of patients who have a diffuse lymphoproliferative process, in the sera of patients who have chronic lymphocytic leukemia with no recognizable effect on the clinical course, in the dermatologic diseases of lichen myxedematosus, pyoderma gangrenosum, and necrobiotic xanthogranuloma, and more often in the peripheral neurologic disorders sensorimotor peripheral neuropathy and chronic inflammatory demyelinating polyneuropathy. MGUS occurs in approximately 5% to 10% of adult patients who have chronic idiopathic axonal polyneuropathy (CIAP), which represents a sixfold increase over the rate found in the general population .
The typical clinical presentations of the MGUS protein classes associated with chronic polyneuropathy are similar and usually begin in the sixth decade of life, progressing in a slow, insidious pattern as distal symmetric sensorimotor polyneuropathy. Sensory deficits begin in the toes and extend up the lower limbs to a greater extent than in the upper extremities. Muscle stretch reflexes are globally diminished or absent with almost universal sparing of cranial nerve function. Paresthesia, ataxia, and pain may be significant but seldom lead to complete inability to walk. The neuropathy in MGUS tends to be more relentlessly progressive than the typical relapsing–remitting course of chronic inflammatory demyelinating polyradiculoneuropathy (CIDP). Differentiating a patient who has MGUS from one who has another plasma cell disorder is difficult on clinical grounds alone. Use of laboratory and additional diagnostic study findings are often helpful in this regard. The recommended diagnostic criteria in patients who have suspected MGUS are summarized in Table 2 . The initial studies to include are a compete blood count, serum creatinine, and serum calcium. If irregularities are identified in any of these tests, a plain film radiograph bone survey including long bones (the humerus and femur bilaterally) is done. A bone marrow aspirate and biopsy are recommended if the M-protein value is greater than or equal to 1.5 g/dL, if an IgA or an IgM MGUS is identified, or if a patient who has an abnormal serum free light chain (FLC) ratio is encountered .
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


