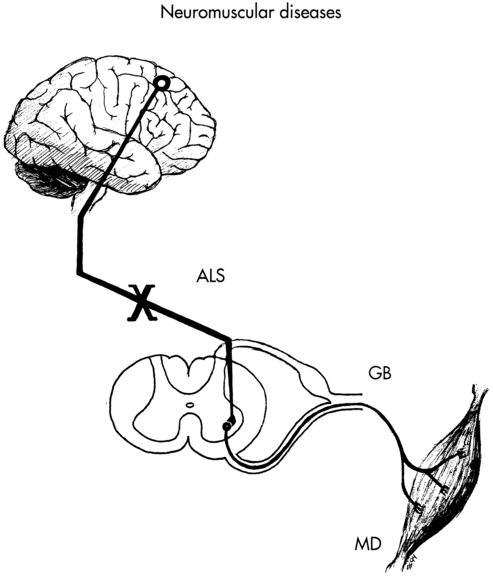
ANN HALLUM, PT, PhD and DIANE D. ALLEN, PT, PhD After reviewing this chapter the student or therapist will be able to: 1. Describe the basic pathology and medical treatment of amyotrophic lateral sclerosis, Guillain-Barré syndrome, and Duchenne muscular dystrophy. 2. Describe the current goals and interventions for each condition. 3. Describe the “safe” exercise windows related to disuse atrophy and exercise (overwork) damage. 4. Be able to apply intervention concepts discussed in this chapter to other neuromuscular diseases. Neuromuscular diseases encompass disorders of upper or lower motor nerves or the muscles they innervate. This chapter traces the connections among the central nervous system (CNS), peripheral nervous system (PNS), and musculoskeletal system through the disordered functioning associated with three neuromuscular diseases: amyotrophic lateral sclerosis (ALS), which damages upper and lower motor neurons; Guillain-Barré syndrome (GBS), which compromises lower motor neurons and the PNS; and Duchenne muscular dystrophy (DMD), which affects the muscles themselves. To review the normal connections, upper motor neurons originate in the motor cortex of the brain (Betz cells). Axons from these upper motor neurons descend by means of the corticobulbar and corticospinal tracts to synapse with lower motor neurons in the brain stem (neurons of the cranial nerves with motor functions) and spinal cord (anterior horn cells or alpha motor neurons). Simultaneously, corticobulbar tract fibers innervate neurons originating within the brain stem and descending through the spinal cord to provide additional input to lower (alpha) motor neurons. Axons from the lower motor neurons within both the brain stem and spinal cord run within the peripheral nerves, which include motor and sensory fibers, to synapse with muscle fibers. The muscle fibers respond to excitation by contracting. Depending on the site of the pathology, neuromuscular diseases can be classified as neurogenic or myopathic. ALS and GBS are neurogenic disorders; DMD is a primary myopathy (Figure 17-1). In considering the movement dysfunction associated with these diseases, strength and endurance are most affected, with flexibility deficits resulting from these.1 All three disorders decrease a person’s ability to generate force in the affected muscles, with weakness as a primary symptom. Loss of muscle strength can lead to speech, swallowing, and respiratory difficulties along with functional limitations. Fatigue is another primary deficit, although the neurogenic disorders tend to result in central fatigue (deficit in ability to recruit motor units) as opposed to the peripheral fatigue of the myopathies (deficit in ability of muscle fibers to contract forcefully).2 Secondary movement problems include loss of range of motion (ROM) in immobile muscles and joints, and pain or muscle spasms. Adaptability, the ability to sense obstacles or changes in the environment and change the course of a movement in response,1 may be affected with the sensory loss in GBS but is not typically a problem in ALS or DMD. The cause of ALS is unknown; however, numerous theories have been proposed. Ninety percent of the cases of ALS are sporadic without a known genetic component; however, most neurodegenerative diseases are now thought to be related to complex protein misfolding disorders. The latest research suggests that ALS and other neurodegenerative disorders are related to TDP-43 proteinopathy.3 Approximately 5% to 10% of the cases seem to have a complex genetic basis coded on ALS1 through ALS8 and other mutations that are associated with frontal lobe dementias. Twenty percent of genetic causes of ALS are thought to be related to mendelian mutations in the superoxide dismutase–1 (SOD1) gene (ALS1). Other factors considered in the genesis of ALS are vascular endothelial growth factors, toxicity leading to motor neuron death, oxidative stress and mitochondrial dysfunction related to microglial inflammation,4,5 and environmental factors.6 The differential diagnosis for ALS is extensive. The possibility of cervical or lumbar spondylosis, syringomyelia, multiple sclerosis, primary lateral sclerosis, and diseases associated with lower motor neuron pathology, among other diagnoses, needs to be excluded before the diagnosis of ALS is made.7 Currently, no single laboratory test is available to confirm a diagnosis of ALS, although creatine phosphokinase levels are elevated in approximately 70% of patients and tend to be higher in patients with limb onset ALS rather than bulbar onset.8 Genetic testing to identify the mutations in the Cu,Zn SOD1 gene is available when a family history of ALS is present. Other laboratory tests, such as identification of biochemical markers in the blood and cerebrospinal fluid, are used to exclude other neurological diseases. Electromyography (EMG) and nerve conduction studies can be helpful to confirm the presence of widespread lower motor neuron disease without peripheral neuropathy or polyradiculopathy. Neuroimaging studies are used to rule out conditions that may have clinical signs similar to those of ALS.9 Because of the absence of clear laboratory markers of ALS, the clinical diagnosis must be made on the basis of recognition of a pattern of observed and reported symptoms of both upper and lower motor neuron disease and persistent declines in physical functions supported by inclusionary and exclusionary diagnostic testing. Because of the overlap of symptoms with other neuromuscular disorders, misdiagnosis is not uncommon.10 ALS is the most common form of motor neuron disease, with an incidence of approximately three to five cases per 100,000 persons. Mean age at onset is 57 years, with two thirds of patients aged 50 to 70 years old at time of onset.11 Men are affected approximately 1.3 to two times more frequently than are women, although the differences are less with late onset of disease (ages 70+).12 The World Federation of Neurology (WFN) has developed suggested diagnostic criteria (suspected, possible, probable, and definite) for patients with ALS entering clinical research trials. Essentially, a patient with “definite” ALS must show concomitant upper motor neuron and lower motor neuron signs in three spinal regions or in two spinal regions with bulbar signs. Either upper or lower motor signs must also be evident in other regions of the body.13 Exclusionary criteria are oculomotor nerve pathway abnormalities (the oculomotor nerve is spared in ALS), significant movement disorder patterns, sphincter control problems, the presence of sensory and autonomic nervous system (ANS) dysfunction, and cognitive deterioration.14 (Refer to the WFN ALS website [www.wfnals.org] section on ALS education for up-to-date criteria used for clinical studies.) Although a consistent diagnostic criterion for ALS has been the absence of sensory involvement, some evidence exists that there is a progressive functional deficit in sensation, perhaps related to ongoing immobility.15 Similarly, cognitive deficits are considered exclusionary criteria for an ALS diagnosis. However, a small subgroup of patients with both familial and sporadic forms of ALS has been identified as having concomitant evidence of frontotemporal dementia (FTD), showing lower scores on executive cognitive functions, word finding, and phrase length.16 A combination of ALS and FTD suggests a common cause may be possible.17 Because of these findings, therapists should be aware of the possibility of cognitive deficits in their patients with ALS, manifested as a decrement in executive skills such as planning and organization and language problems. Such patients may have more difficulty following through on medication and therapeutic recommendations, and their families may need more support. Unassociated with overall cognitive impairment, some deficits in action knowledge as opposed to object knowledge have been noted in patients with ALS, correlating with atrophy in the motor and premotor cortex.18 Specific cognitive deficits, therefore, may be more common than previously noted. The earliest clinical markers heralding ALS are fasciculations (especially unequivocal fasciculation in the tongue), muscle cramps, fatigue, weakness, and atrophy.13,19 During initial diagnostic visits, patients frequently report to their physicians a profound sense of fatigue or the loss of exercise tolerance.19 Ninety percent of patients report weakness occurring in a striated muscle or group of muscles. Because the onset of ALS is insidious, most patients are not aware of the strength changes, or they have adjusted to the changes until they have difficulty with a functional activity such as tying shoes or climbing stairs. Physical examination usually demonstrates more widespread weakness and atrophy than reported by the patient. By the time most patients report weakness, they have lost approximately 80% of their motor neurons in the areas of weakness. This demonstrates the plasticity of the nervous system and its drive to adapt to meet functional goals. The weakness spreads over time to include musculature throughout the body. Succeeding symptoms of weakness in other muscles depends on the continued loss of motor neurons to the 20% threshold needed for perception of weakness.20,21 A typical, but not absolute, pattern of motor progression is early distal involvement followed by proximal limb involvement. In some cases bulbar symptoms herald the onset of ALS, but bulbar symptoms more commonly occur later in the disease. Flexor muscles tend to be weaker than extensor muscles.22 Although the atrophy and weakness component of ALS is most obvious, 80% or more of patients show early clinical evidence of pyramidal tract dysfunction (e.g., hyperreflexia in the presence of weakness and atrophy, spasticity, and Babinski and Hoffmann reflexes).13 Although in some cases the upper motor neuron signs may be absent clinically, Chou23 has shown on autopsy that significant involvement may be present despite the lack of clinical evidence. The pattern of ALS onset is highly varied, with several patterns identified by primary area of onset. Lower-extremity onset is slightly more common than upper-extremity onset, which is more common than bulbar onset. Some patients show initial symptoms in distal musculature of upper and lower extremities. A significant diagnostic feature of the pattern of disease is the asymmetry of the weakness and the sparing of some muscle fibers even in highly atrophied muscles. For example, a patient may have weakness of the right intrinsics and shoulder musculature or weakness of the left anterior tibial muscles. Bulbar symptoms are presaged by tongue fasciculations and weakness, facial and palatal weakness, and swallowing difficulties, which result in dysphagia and dysarthria. Pseudobulbar palsy is sometimes present in ALS, manifested by spontaneous laughing or crying unrelated to the situation.24 Despite the pattern of onset, however, the eventual course of the illness is similar in most patients, with an unremitting spread of weakness to other muscle groups leading to total paralysis of spinal musculature and muscles innervated by the cranial nerves. Death is usually related to respiratory failure.25 In a longitudinal study using monthly questionnaires, direct patient interviews, record reviews, physician interviews, and family member interviews, Brooks and colleagues20 followed 702 patients with ALS. Their findings suggest that spread of neuronal degeneration occurred more quickly to adjacent areas than to noncontiguous areas. The spread to adjacent areas was more rapid at the brain stem, cervical, and lumbar regions. Limb involvement after bulbar onset was more aggressive in men than in women.20 One study focused on developing methods to assess the natural history of the progression of ALS so that medical and supportive treatment planning and interventions could be instituted.26 Hillel and colleagues27 have developed the ALS severity scale for rapid functional assessment of disease stage. Their 10-point ordinal scale allows clinicians and therapists to score patients in four categories: speech, swallowing, and lower-extremity and upper-extremity function (Box 17-1). BOX 17-1 Adapted with permission from Hillel AD, Miller RM, Yorkston K, et al: Amyotrophic lateral sclerosis severity scale. Neuroepidemiology 8:142, 1989. A five-point scale of severity is currently being used in ALS clinical drug trials. Patients in stage 1 (mild disease) have a recent diagnosis and are functionally independent in ambulation, activities of daily living (ADLs), and speech. Stage 2 (moderate) identifies patients with mild deficits in function in three regions or a moderate to severe deficit in one region and mild or normal function in two other regions. Stage 3 (severe) defines patients who need assistance because of deficits in two or three regions; for example, the patient needs assistance to walk or transfer, needs help with upper-extremity activities, and/or is dysarthric or dysphasic. Stage 4 identifies patients with nonfunctional movement of at least two regions and moderate or nonfunctional movement of a third area. Stage 5 is death.22 (See Brooks and colleagues14 and Pradas and colleagues28 for information on the natural history of ALS and its importance in the design of clinical treatment trials.) Along with the primary impairments of weakness and fatigue affecting body structure and function in ALS, patients also have progressive limitations in activity and participation.29 Activity limitations result in gradual loss of independence in community and then household tasks. Mechanical and electronic adaptive devices can help extend independence in some ADLs past the initial strength losses. Participation limitations result in progressive isolation from the community and family unless extraordinary efforts persist to retain a communication system at home and through electronic media. In almost all cases ALS progresses relentlessly and leads to death from respiratory failure. The rate of progression seems to be consistent for each patient but varies considerably among patients. Patients with an initial onset of bulbar weakness (dysarthria, dysphagia) and respiratory weakness (dyspnea) tend to have a more rapid progression to death than patients whose weakness begins in the distal extremities.30 Death usually follows within 2 to 4 years after diagnosis, with a small number of patients living for 15 to 20 years.10 Years of survival after diagnosis may change as drug therapies are developed.31 In addition, increasing numbers of patients are electing to prolong life with home-based mechanical ventilation as opposed to palliative or comfort care only. ALS has no known cure and minimal effective disease-slowing treatments. Mitchell and Borasio24 have created a table (see Table 2 in their study) that summarizes the results of trials of the many putative ALS-modifying pharmaceuticals. Only riluzole has been approved for treatment of ALS. Riluzole provides very modest improvement over a placebo in both bulbar and limb function, but not in actual strength of muscles.32 The drug extended lifespan an average of 2 to 3 months. The side effects were minimal in some studies, but fatigue and weakness have been noted in 26% and 18% of patients taking riluzole compared with a placebo.33 The popular press has reported on nutritional cures for ALS, including regular use of vitamin E. However, Orrell and colleagues34 found insufficient evidence to support clinical use of vitamin E supplements in ALS as an additive to riluzole treatment or as adjunctive therapy, although no apparent contraindication was found to taking the supplement. Other nutritional and nonpharmaceutical supplements have had some success in animal models of ALS, but this has not yet been confirmed in humans.35 Cannabis has been studied for its effect on spasticity in patients with multiple sclerosis and spinal cord injury. In a study of 131 people with ALS, 13 used cannabis, with reports of reduction in spasticity, pain, and depression.36 Because of the apparent hopelessness of the diagnosis, many physicians, especially those not associated with major medical centers having neuromuscular disease units, do not refer patients with ALS for services, yet few primary care physicians or neurologists have extensive experience in the care of patients and families coping with ALS because of the low incidence of the disease. Yet, referral of patients with ALS to a multidisciplinary clinic typically extends the patient’s lifespan, especially patients with bulbar onset of ALS.25,37 Some patients experience muscle cramps and spasms related to upper motor neuron pathology, and up to 73% of patients complain of pain, typically in the later stages.24 Although most spasms can be relieved with stretching or increased movement, some patients require medications such as quinine or baclofen to relieve symptoms (see Chapter 36 for information on drug therapies). In a review of studies on the treatment of spasticity in ALS, Ashworth and colleagues38 found only one randomized study addressing spasticity: a moderate-endurance exercise regimen decreased spasticity at 3 months after initiation of the program. Stretching and massage may prove helpful for nocturnal muscle cramps.25 Kesiktas and colleagues39 report that in a controlled study of spasticity in patients after spinal cord injury, adding hydrotherapy to a program of medication and exercise decreased severity of spasms and decreased the amount of medication required. A similar response could be hypothesized in patients with ALS. In addition to muscle spasms, patients report nonspecific aching and muscle soreness, probably related to immobility and trauma to paralyzed muscles during caregiving procedures. However, many patients do not receive adequate pain medication, or the pain is not controlled by the medication taken.40 A Cochrane review in 2008 found no randomized or quasi-randomized controlled trials of drug therapy for pain in ALS, although several case series reported the use of acetaminophen, nonsteroidal antiinflammatory drugs (NSAIDs), or opioids.41 Careful administration of medications such as baclofen, tizanidine, dantrolene sodium, and diazepam is useful for some patients with spasticity. Because each has a different action and side effects, the medications may have to be adjusted to find the right dosage and combination. In some patients with severe cramping, botulinum toxin injections might be helpful, but they must be carefully administered to prevent further weakness. Because many patients have compromised respiratory function, the physician must take great care when prescribing pain medication, especially opiates, which are often used when antispasmodics or antiinflammatory pain medications no longer work.25 Patients should be instructed to keep a daily reporting log of the effectiveness of the medication so that the dosage can be adjusted if necessary. Dysphagia, a difficulty swallowing liquids, foods, or saliva, accounts for considerable misery in the patient with advanced ALS, and it must be dealt with aggressively. Patients with dysphagia have both nutritional and swallowing problems associated with weakness of the lips, tongue, palate, and mastication muscles.42 As the progressive loss of swallowing develops, patients are also at extreme risk for aspiration. Most patients with dysphagia also have severe problems with management of their saliva (sialorrhea). If a patient has difficulty transporting saliva back to the oropharynx for swallowing, choking and drooling are common.43 This condition is disconcerting to the affected person, who must constantly wipe the mouth or have someone do it for him or her. In addition, secretions are often thickened because of dehydration. With pooling of the thickened saliva, the possibility of aspiration is increased. Viscosity of saliva can best be treated by hydration and, in some cases, pharmaceuticals. Drugs, such as decongestants, antidepressant drugs with anticholinergic side effects, and atropine-type drugs, can help control the amount of saliva, provided the patient is well hydrated.44 In extreme cases, various surgical procedures such as ligation of the salivary gland ducts, severing the parasympathetic supply to the salivary glands, and excision of the salivary glands have been used effectively.45 Newer treatments to decrease excessive secretions are radiotherapy and botulinum A toxin injections into salivary glands.46 Although dietary treatment is not known to be effective in changing the course of the disease, a nutritious diet to meet caloric, fluid, vitamin, and mineral needs must be maintained. Seventy-three percent of patients with ALS have difficulty bringing food to the mouth, making them dependent on others for their dietary needs. Because of the time it takes to be fed, many patients decrease their intake. All patients with dysphagia should be referred for a dietary consultation to determine the choice and progression of solid and liquid foods and supplements.47 Appel and colleagues47 describe nutritional plans to maintain nutrition and hydration in patients with motor neuron diseases. Patients with bulbar symptoms and severe dysphagia who are no longer able to consume nutrients orally because of motor control problems and recurrent aspiration may need a percutaneous endoscopic gastrostomy (PEG) for feeding, depending on the patient’s wishes for long-term care. Some evidence exists that the PEG should be performed early in the disease process to prevent severe weight loss and aspiration.48 Although a PEG does not appreciably lengthen survival time,49 patients may have less fear of choking or aspiration. Receiving nourishment from a PEG does not prevent the person from taking food orally if desired. Dysarthria, impairment in speech production, is the result of abnormal function of the muscles and nerves associated with coordinated functions of the tongue and lips, larynx, soft palate, and respiratory system. Speech impairments are the initial symptom in most patients with bulbar involvement. Speech intelligibility is compromised by hypernasality, abnormalities of speed and cadence of speech, and reduced vocal volume. Speech is further compromised by inadequate breath volumes for normal phrasing. A possible option to help patients with severe hypernasality is a palatal lift prosthesis to augment velopharyngeal function.50,51 Because little can be done medically to delay the loss of speech control, early referral to a speech therapist is essential. Numerous augmentative and alternative communication systems are now available, the simplest being voice amplification systems or homemade point boards and computer-based head or eye tracking text-to-speech systems that can be modified as the patient status changes. The type of communication system should be chosen with awareness of the patient-caregiver environment.52 Progressive respiratory failure is the primary cause of death in ALS patients. Respiratory failure is related to primary diaphragmatic, intercostal, and accessory respiratory muscle weakness.53 Respiratory failure should be anticipated and discussed early following the diagnosis of ALS so that patients and their caregivers can express their wishes and develop an advanced directive for care in the terminal phase of the disease.54 Physiological tests used to indicate respiratory dysfunction include vital capacity, sniff nasal pressure, and nocturnal oximetry.10 Clinical signs of increased respiratory dysfunction are dyspnea with exertion or lying supine; hypoventilation; weak or ineffective cough; increased use of auxiliary respiratory muscles; tachycardia (also a sign of pulmonary infection with fever and tachypnea); changes in sleep pattern; daytime sleepiness and concentration problems; mood changes; and morning headaches.55 In early stages of patient care, physical therapists (PTs) may help manage respiratory dysfunction by providing postural drainage with cough facilitation (suctioning if necessary), especially during acute respiratory illnesses. The patient and care providers should also be taught breathing exercises, chest stretching, and incentive spirometry techniques, as well as postural drainage techniques if the caregivers are prepared to provide such support. Although breathing exercises consisting of resisted inspiratory muscle training can facilitate functional respiration, even practicing unresisted breathing for 10 minutes three times a day has been shown to result in improved function.56 An assessment of the home environment is imperative to identify sleeping positions and energy conservation techniques that can be incorporated into the patient’s daily life. As respiratory symptoms increase, oxygen at 2 L/min or less can be used intermittently at home. When hypoventilation with a decline in oxygen saturation becomes common during sleep, resulting in morning confusion and irritability, patients have the option to initiate noninvasive, positive-pressure ventilation (NIV) such as bilevel positive airway pressure (BiPAP). BiPAP, which provides greater inspiratory pressure than expiratory pressure to decrease the effort of breathing, can be administered by either mask or contoured nasal delivery systems. Some evidence indicates that early use of NIV can increase survival time by several months and increase quality of life.57 When a patient can no longer benefit from NIV, a decision must be made about initiating ventilation by tracheostomy or palliative care.58 (See also Miller and colleagues59 for an excellent discussion of practice parameters in the decision-making process related to ventilatory support.) Although in the initial stages of ALS most patients indicate they would not want prolonged respirator dependence at home, patients may change their minds as they adapt to the disease restrictions.60 A small study of patients who started tracheostomy intermittent positive-pressure ventilation (TIPPV) demonstrated increased long-term survival (2 to 64 months).54 In another series of 70 patients on long-term TIPPV, 50% of the patients were living after 5 years; however, 11.4% of these patients had entered a “locked-in state in which they were unable to communicate in any manner.”61 Decisions about long-term respirator use should be made by the patient and involved family members or partners, with input from the interdisciplinary team caring for the patient. Discussions of preferred long-term care options should be revisited as the patient’s condition changes. If a patient decides that home ventilation is a reasonable option, those involved in the decision should visit another patient who is using in-home mechanical ventilation, if possible. Because the decision for home mechanical ventilation (HMV, NIV, or TIPPV) also affects the life of the patient’s spouse, children, and extended family who may be responsible for some aspects of home care, or whose lives may be affected by the presence of in-home nurses or attendants, the decision for HMV should not be taken lightly. Extensive preparation, ongoing support, and respite options for caregivers are necessary if HMV is to be successful. Success of HMV also depends on such variables as third-party payment for home care equipment and nurse or attendant staffing, working status of the partner or spouse, age and physical fitness of the spouse and children, pre-ALS family psychosocial interactions, and financial factors. HMV should be viewed as long term, often extending for more than 1 year. Initiation of HMV results in a reasonable perceived quality of life for the patient, yet caregivers report that their quality of life may be lower than the patient’s because of the burden of care that must be provided.62 With chronic respiratory insufficiency, the patient and family must be involved in the long-term care decisions related to instituting mechanical assistance under either emergency situations or in response to gradual deterioration. This discussion should occur before the patient develops respiratory failure. Acute respiratory failure can be frightening, and few patients or family members are prepared to forego intubation and artificial ventilation during the emergency. Patients and caregivers should understand that not making a decision about mechanical ventilation, noninvasive or invasive, is a decision to support mechanical ventilation.63 Physicians and health care workers who work with the patient and family must be aware of their own feelings and beliefs about prolonging life. For example, a healthy physician or therapist who values control and an active lifestyle may envision a life on a ventilator as intolerable and pass that value on to the patient, who may or may not have the same needs. The patient’s decision, or change in decision, must be respected by the medical team involved in care.64 In medical centers that use a team approach, patients and families may find support by meeting with counselors or peers with ALS who are making or have made decisions about long-term ventilator care. Perhaps because of the multitude of issues to consider when managing the impairments and limitations associated with ALS, evidence suggests that patients treated by a specialized ALS multidisciplinary team fare better than do those treated by single-source providers,65 or in general neurology clinics.33 A Cochrane review of the evidence for multidisciplinary care advantages in this population concluded that the evidence is of low quality, so far, with no controlled trials identified.66 Whether administered through an ALS-specific team or not, therapeutic management will necessitate examination of the patient’s current status, evaluation of the deficits in relation to patient preferences and needs, and establishment of a plan based on mutually determined and realistic goals. The rate of the patient’s disease progression, the areas and extent of involvement, and the stage of illness must be considered. A patient at the initial stages will have different needs than a patient at later stages who has chosen NIV or tracheostomy ventilation that may extend life span at a markedly reduced mobility level. The goal at all stages is to optimize health and increase the quality of life. With guidance and environmental adaptations, patients with slowly progressing weakness may be able to continue many of their ADLs for an extended number of years. In the final stages of the disease, when the patient is bedridden, programs to increase strength or endurance are not appropriate, and interventions such as stretching may not effectively control contracture development. However, patients may still benefit from positioning and range-of-motion (ROM) exercises to decrease muscle and joint pain related to immobility. The prescription of assistive devices and training of caregivers will also be needed. The efficacy of therapeutic interventions will be related to the timing of interventions, the motivation and persistence of the patient in carrying out the program, and support from family members or caregivers.67 Objective documentation of outcome measures will help justify the usefulness of therapeutic interventions at all stages of this disease. If possible, before the patient’s initial visit, the therapist should contact the patient and request that he or she keep an activity log for several days. If an early contact is not possible, the therapist can assign that task during the initial session. The log should include 15-minute time increments in which the patient or caregiver can record what she or he was doing during a specific period. The log should also indicate whether the patient was experiencing fatigue or pain during the activity and how the patient perceived her or his respiratory status. An example of an activity log and how it is used is shown in Figure 17-2. The sense of fatigue with repetitive muscle activity or functional activity should be specifically tracked by the patient. Weakness will be the primary deficit, with other problems following depending on the location of strength loss. Muscle weakness and the experience of fatigue may be independent measures of ALS pathology, however.68 Although weakness may affect balance during gait, patients with ALS have not shown deficits in postural control during quiet stance despite significant paresis or tone changes, possibly because sensation is relatively preserved.69 The therapist’s examination will vary depending on the patient’s situation29; however, a typical initial assessment may include the following: TABLE 17-1 COMMON PHYSICAL FINDINGS IN BULBAR AMYOTROPHIC LATERAL SCLEROSIS Modified with permission from Hillel AD, Miller RM: Bulbar amyotrophic lateral sclerosis: patterns of progression and clinical management. Head Neck 11:51–59, 1989. Copyright 1989. Reprinted by permission of John Wiley & Sons, Inc. Brinkmann and colleagues70 identify standards for assessment of patients with ALS in clinical trials. The review and description of standardized methods for performing recommended tests and measurements is extremely valuable for any therapist assessing and treating patients with ALS. Intervention goals and the recommended exercise and activity program designed by PTs or OTs must be based on the patient’s personal goals. Goals are often a difficult area for therapists to discuss with the patient because the disease is progressive despite intervention. Patients, therapists, and physicians commonly assume that because nothing can be done to “cure” the disease, not making additional demands on a patient who is already coping with daily loss is somehow kinder. Some believe that exercise programs may create false hopes that exercise will delay progression. Others believe that exercise will hasten progression.71 The literature on rehabilitation in neuromuscular disorders, however, suggests that patients with ALS can benefit from carefully designed exercise and activity programs. Active participation in determining goals for therapy can provide the patient and the family with some sense of control over a difficult situation.7 The general, broad goals for both patient and therapist are related to maintaining maximal independence in daily living and a positive quality of life for as long as possible. More specific therapeutic goals are (1) maintenance of mobility and independent functioning, to include safe mobility for patient and caregiver; (2) maintenance of maximal muscle strength and endurance within limits imposed by ALS; (3) prevention and minimization of secondary consequences of the disease, such as contractures, thrombophlebitis, decubitus ulcers, and respiratory infections7,67; (4) management of energy conservation techniques and respiratory comfort; (5) determination of adaptive equipment needs to include mobility, self-help and feeding devices, augmentative communication units, and hygiene equipment that supports both patient and caregiver7; and (6) eliminating or preventing pain.72 To prevent more rapid functional loss than expected from the natural history of the disease, both the patient and therapist must delicately balance the level of activity between the extremes of inadequate exercise and excessive exercise. Exercise has been recommended for the general public for its many benefits.73 Inadequate exercise may result in loss of strength and endurance from disuse, as well as secondary problems such as loss of ROM, muscle cramping, and pain. Excessive exercise may result in excessive fatigue and consequent inability to perform ADLs during recovery periods. Overuse injury with excessive strengthening exercise may also lead to unnecessary pain and loss of strength. The next two sections review the evidence for the optimum amount of activity or exercise. Because ALS is a disease of older adults, patients may not have maintained their aerobic fitness or muscle strength before the onset of their neuromuscular problem. Newly diagnosed patients also commonly report that they had markedly decreased their activity level in the months before diagnosis because of a sense of fatigue or increasing clumsiness from increasing weakness. If the patient had led a sedentary lifestyle before diagnosis, the additional decrease in activity level after the onset of ALS can lead quickly to marked cardiovascular deconditioning and disuse weakness. The disuse weakness lowers muscle force production and reduces muscle endurance.74 Anecdotal evidence that muscle activity or overwork exercise can lead to a loss of muscle strength has been reported since the poliomyelitis epidemic of the 1940s and 1950s.75 During that epidemic, physicians and therapists noted that patients with poor- and fair-grade muscles who exercised repeatedly or with heavy resistance after reinnervation often lost the ability to contract the muscle at all76 (see Chapter 35). Controlled testing of this observation suggests that overwork damage occurs in mostly denervated muscles, not in all muscles. Reitsma77 noted that vigorous exercise damaged muscles in rats if less than one third of motor units were functional. If more than one third of the motor units remained, exercise led to hypertrophy. An additional mechanism of potential overwork damage is inhibition of the collateral sprouting of intact axons to innervate “orphaned” muscle fibers when other axons degenerate. Yuen and Olney78 provided evidence that collateral sprouting of intact axons can partially reinnervate orphaned muscle fibers in ALS. In a rat model, highly intensive activity reduced the ability of adjacent axons to sprout after fewer than 20% of intact motor units remained.79 In contrast, vigorous exercise in a mouse model had no adverse effect on the course of ALS.80 Lui and Byl81 systematically reviewed the literature reporting exercise effects in animal models of ALS and calculated an effective size of 1.39 (where numbers over 0.8 are considered large) in favor of exercise. The few negative effects they noted were associated with either very-high–intensity exercise or a slow rate of exercise (slower than usual activity for animals when unrestricted in activity). In addition to generic overwork, evidence exists that repeated maximal eccentric contractions may specifically damage even normal muscle fibers, resulting in muscle weakness of several weeks’ duration.82 Although normal muscle eventually adapts to repeated eccentric exercise, whether the reparative effect is possible in patients with neuromuscular diseases is uncertain. Aboussouan55 reviews some of the specific mechanisms of exercise intolerance in neuromuscular diseases, including mitochondrial dysfunction, abnormal muscle metabolism, impaired muscle activation, and central activation failure. Many researchers have expressed concern about the possible relation between high-resistance exercise and muscle fiber degeneration in humans with motor neuron disease.83,84 Because of the concerns about damage from stressing substantially denervated muscles, Sinaki and Mulder85 published recommendations in 1978 that patients with ALS not engage in any vigorous exercise and focus instead on exercise associated with walking and daily activities. On the other hand, McCrate and Kaspar86 review the possible mechanisms by which exercise protects nerves from more rapid degeneration. Evidence regarding the positive benefits of exercise in ALS has been accumulating, with fewer adverse effects than some expected. Sanjak and colleagues87 reported that muscle damage does not necessarily result from resistance exercise testing or training, although fatigue occurs more easily during both anaerobic and aerobic exercise. Milner-Brown and Miller88 found that mild progressive resistance exercise was helpful in neuromuscular disorders if the patient had muscle strength in the good (4/5) to normal (5/5) range. They determined that patients should begin their exercise program early because strength training of muscles with less than 10% of normal function was generally not effective. Aitkens and colleagues89 noted strength gains of 4% to 20% without deleterious effects after a 12-week program of moderate-resistance (30% of maximum isometric force) exercises in patients with slowly progressive neuromuscular diseases. Kilmer and colleagues,90 in the same population, found no additional advantage to high-resistance training (12 weeks of exercise using the maximum isometric force the individual was able to lift 12 times) and noted evidence of overwork in some subjects. In a case report of a patient with ALS, strengthening 6 days a week for 10 weeks with proprioceptive neuromuscular facilitation (PNF) patterns using maximal resistance applied manually or with tubing resulted in strengthening of 14 muscle groups out of 18 with no adverse effects.91 Aksu and colleagues92 compared a supervised versus home exercise protocol in 26 ambulatory ALS patients. They noted that supervised breathing exercises, stretching, manually applied resistance exercise with PNF, and functional mobility training 3 days a week for 8 weeks resulted in small gains in function in the first 4 weeks and a slower decline over the subsequent 10 months compared with home-based breathing, stretching, and active ROM exercises. The groups were not randomly allocated but were not significantly different in the measured variables at baseline.92 In a randomized controlled trial, Drory and colleagues93 assigned 25 patients with ALS to a group continuing their normal daily activities or a group participating in a moderate daily program of exercise individualized for each patient. The primary exercise focus was to have muscles of the trunk and limbs work against “modest” loads while undergoing significant shortening (not lengthening or eccentric contractions). The exercises were completed twice daily for 15 minutes at home with phone contact by the treating therapist every 14 days. Data were evaluated for 3 and 6 months after initial assessment. All patients showed continued disease progression; however, in all cases, at the 6-month assessment patients who exercised showed positive effects in maintenance of muscle strength, less fatigue, less spasticity, less pain, and higher functional ratings.93 In another randomized controlled trial, moderate load and moderate-intensity resistance exercises prescribed individually to patients with ALS in the early stages resulted in significantly less decline in function, small improvements in strength, and no reported adverse effects, compared with patients who performed stretching exercises alone.94 A Cochrane review designated the quality of the Drory and colleagues (2001) study as “fair” and the Dal Bello-Haas and colleagues94 study as “adequate.”95 Table 17-2 summarizes some of the studies of strength training in neuromuscular diseases. TABLE 17-2 SUMMARY OF STRENGTH TRAINING STUDIES IN NEUROMUSCULAR DISEASES
Neuromuscular diseases
Amyotrophic lateral sclerosis
Pathology and medical diagnosis
Clinical presentation
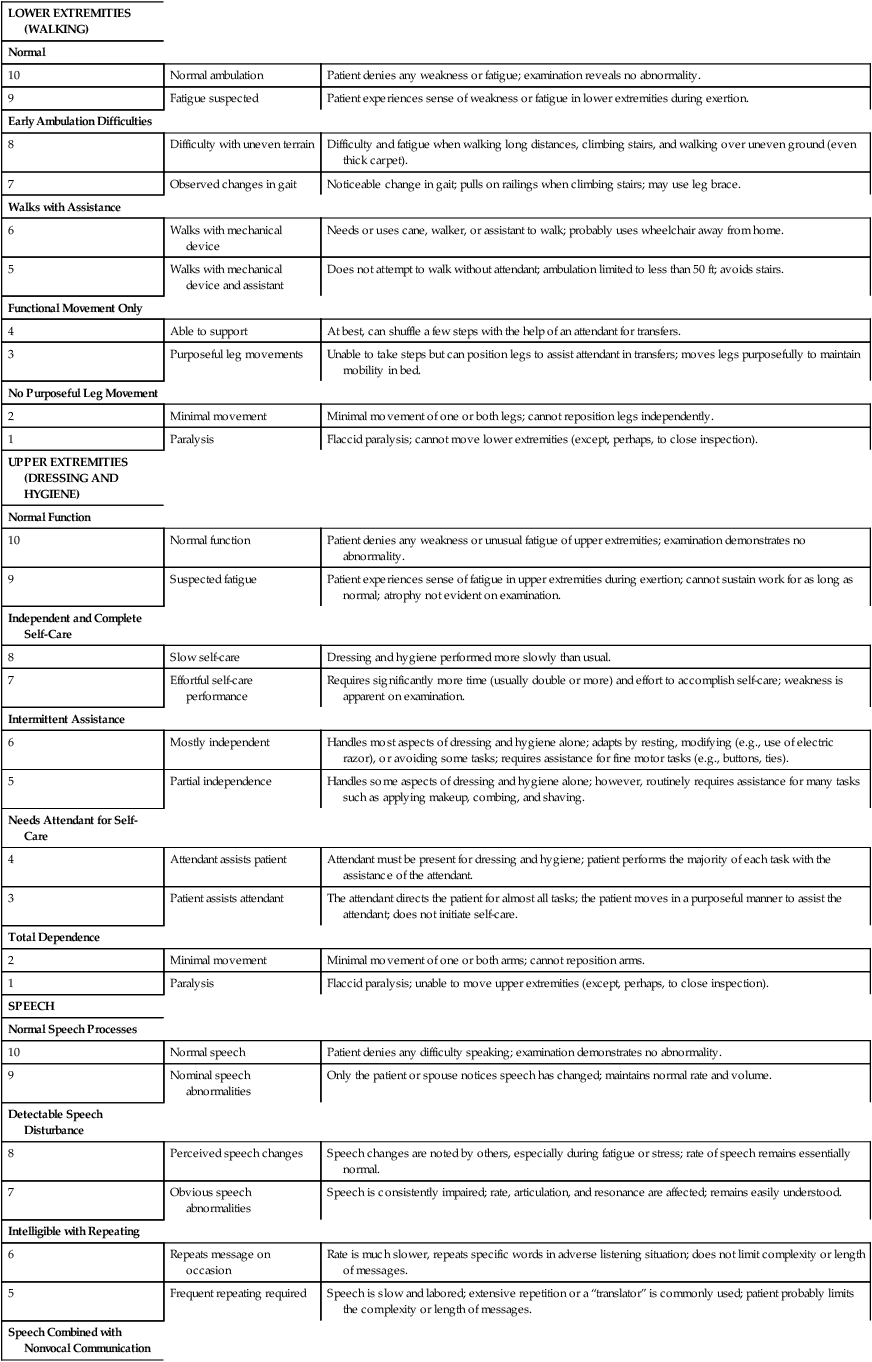
 AMYOTROPHIC LATERAL SCLEROSIS SEVERITY SCALE: LOWER EXTREMITY, UPPER EXTREMITY, SPEECH, SWALLOWING
AMYOTROPHIC LATERAL SCLEROSIS SEVERITY SCALE: LOWER EXTREMITY, UPPER EXTREMITY, SPEECH, SWALLOWING
LOWER EXTREMITIES (WALKING)
Normal
10
Normal ambulation
Patient denies any weakness or fatigue; examination reveals no abnormality.
9
Fatigue suspected
Patient experiences sense of weakness or fatigue in lower extremities during exertion.
Early Ambulation Difficulties
8
Difficulty with uneven terrain
Difficulty and fatigue when walking long distances, climbing stairs, and walking over uneven ground (even thick carpet).
7
Observed changes in gait
Noticeable change in gait; pulls on railings when climbing stairs; may use leg brace.
Walks with Assistance
6
Walks with mechanical device
Needs or uses cane, walker, or assistant to walk; probably uses wheelchair away from home.
5
Walks with mechanical device and assistant
Does not attempt to walk without attendant; ambulation limited to less than 50 ft; avoids stairs.
Functional Movement Only
4
Able to support
At best, can shuffle a few steps with the help of an attendant for transfers.
3
Purposeful leg movements
Unable to take steps but can position legs to assist attendant in transfers; moves legs purposefully to maintain mobility in bed.
No Purposeful Leg Movement
2
Minimal movement
Minimal movement of one or both legs; cannot reposition legs independently.
1
Paralysis
Flaccid paralysis; cannot move lower extremities (except, perhaps, to close inspection).
UPPER EXTREMITIES (DRESSING AND HYGIENE)
Normal Function
10
Normal function
Patient denies any weakness or unusual fatigue of upper extremities; examination demonstrates no abnormality.
9
Suspected fatigue
Patient experiences sense of fatigue in upper extremities during exertion; cannot sustain work for as long as normal; atrophy not evident on examination.
Independent and Complete Self-Care
8
Slow self-care
Dressing and hygiene performed more slowly than usual.
7
Effortful self-care performance
Requires significantly more time (usually double or more) and effort to accomplish self-care; weakness is apparent on examination.
Intermittent Assistance
6
Mostly independent
Handles most aspects of dressing and hygiene alone; adapts by resting, modifying (e.g., use of electric razor), or avoiding some tasks; requires assistance for fine motor tasks (e.g., buttons, ties).
5
Partial independence
Handles some aspects of dressing and hygiene alone; however, routinely requires assistance for many tasks such as applying makeup, combing, and shaving.
Needs Attendant for Self-Care
4
Attendant assists patient
Attendant must be present for dressing and hygiene; patient performs the majority of each task with the assistance of the attendant.
3
Patient assists attendant
The attendant directs the patient for almost all tasks; the patient moves in a purposeful manner to assist the attendant; does not initiate self-care.
Total Dependence
2
Minimal movement
Minimal movement of one or both arms; cannot reposition arms.
1
Paralysis
Flaccid paralysis; unable to move upper extremities (except, perhaps, to close inspection).
SPEECH
Normal Speech Processes
10
Normal speech
Patient denies any difficulty speaking; examination demonstrates no abnormality.
9
Nominal speech abnormalities
Only the patient or spouse notices speech has changed; maintains normal rate and volume.
Detectable Speech Disturbance
8
Perceived speech changes
Speech changes are noted by others, especially during fatigue or stress; rate of speech remains essentially normal.
7
Obvious speech abnormalities
Speech is consistently impaired; rate, articulation, and resonance are affected; remains easily understood.
Intelligible with Repeating
6
Repeats message on occasion
Rate is much slower, repeats specific words in adverse listening situation; does not limit complexity or length of messages.
5
Frequent repeating required
Speech is slow and labored; extensive repetition or a “translator” is commonly used; patient probably limits the complexity or length of messages.
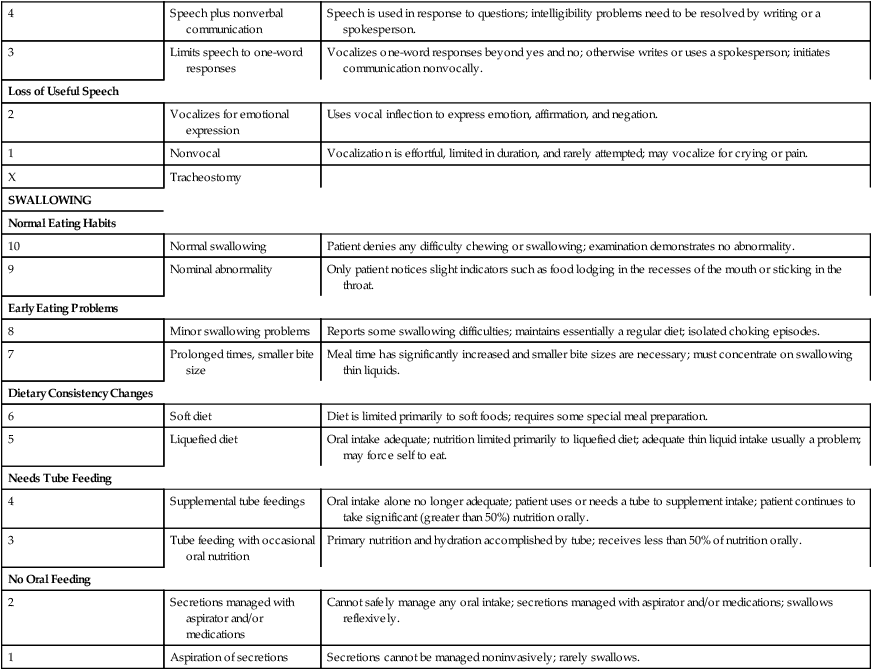
Speech Combined with Nonvocal Communication
4
Speech plus nonverbal communication
Speech is used in response to questions; intelligibility problems need to be resolved by writing or a spokesperson.
3
Limits speech to one-word responses
Vocalizes one-word responses beyond yes and no; otherwise writes or uses a spokesperson; initiates communication nonvocally.
Loss of Useful Speech
2
Vocalizes for emotional expression
Uses vocal inflection to express emotion, affirmation, and negation.
1
Nonvocal
Vocalization is effortful, limited in duration, and rarely attempted; may vocalize for crying or pain.
X
Tracheostomy
SWALLOWING
Normal Eating Habits
10
Normal swallowing
Patient denies any difficulty chewing or swallowing; examination demonstrates no abnormality.
9
Nominal abnormality
Only patient notices slight indicators such as food lodging in the recesses of the mouth or sticking in the throat.
Early Eating Problems
8
Minor swallowing problems
Reports some swallowing difficulties; maintains essentially a regular diet; isolated choking episodes.
7
Prolonged times, smaller bite size
Meal time has significantly increased and smaller bite sizes are necessary; must concentrate on swallowing thin liquids.
Dietary Consistency Changes
6
Soft diet
Diet is limited primarily to soft foods; requires some special meal preparation.
5
Liquefied diet
Oral intake adequate; nutrition limited primarily to liquefied diet; adequate thin liquid intake usually a problem; may force self to eat.
Needs Tube Feeding
4
Supplemental tube feedings
Oral intake alone no longer adequate; patient uses or needs a tube to supplement intake; patient continues to take significant (greater than 50%) nutrition orally.
3
Tube feeding with occasional oral nutrition
Primary nutrition and hydration accomplished by tube; receives less than 50% of nutrition orally.
No Oral Feeding
2
Secretions managed with aspirator and/or medications
Cannot safely manage any oral intake; secretions managed with aspirator and/or medications; swallows reflexively.
1
Aspiration of secretions
Secretions cannot be managed noninvasively; rarely swallows.
Medical prognosis
Medical management
Muscle spasms and pain
Dysphagia
Dysarthria
Respiratory management
Therapeutic management of movement dysfunction associated with ALS
Examination

 Example of a log for monitoring activity level of patients with amyotrophic lateral sclerosis.
Example of a log for monitoring activity level of patients with amyotrophic lateral sclerosis.
 Review of the patient’s medical and activity records, especially time since diagnosis, time course of disease progression to date, current medications, concurrent medical issues, current activities and participation and tolerance for them.
Review of the patient’s medical and activity records, especially time since diagnosis, time course of disease progression to date, current medications, concurrent medical issues, current activities and participation and tolerance for them.
 Assessment of functional activity level (using a standardized test or assessment tool whenever possible) to include, as appropriate: transfers, gait, upper-extremity function, postural control, and assistive devices; suggested tools include the ALS functional rating scale (ALSFRS),70 the ALS severity scale (ALSSS),27 timed walk test, or Purdue Pegboard.70
Assessment of functional activity level (using a standardized test or assessment tool whenever possible) to include, as appropriate: transfers, gait, upper-extremity function, postural control, and assistive devices; suggested tools include the ALS functional rating scale (ALSFRS),70 the ALS severity scale (ALSSS),27 timed walk test, or Purdue Pegboard.70
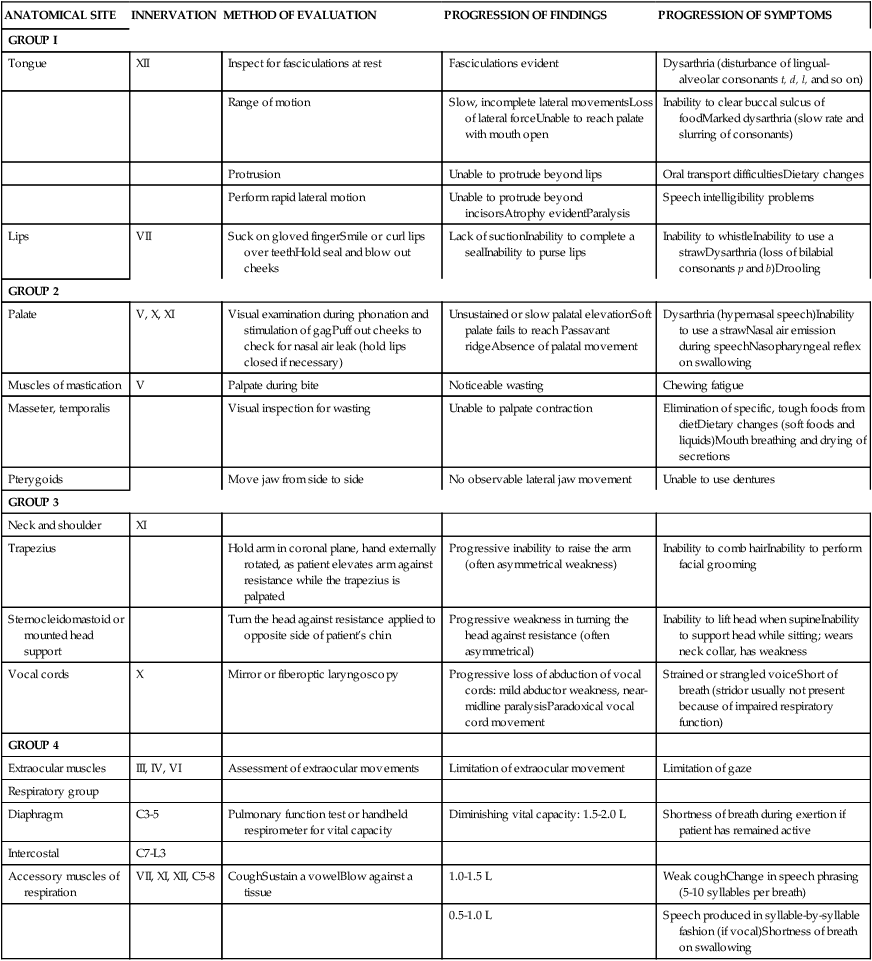
 Assessment of bulbar and respiratory function. (For an in-depth evaluation of bulbar function, the patient should be referred to an ear, nose, and throat clinic or communications disorders clinic unless full evaluation is available in a comprehensive ALS clinic. See Table 17-1 for bulbar and respiratory evaluation suggestions.)
Assessment of bulbar and respiratory function. (For an in-depth evaluation of bulbar function, the patient should be referred to an ear, nose, and throat clinic or communications disorders clinic unless full evaluation is available in a comprehensive ALS clinic. See Table 17-1 for bulbar and respiratory evaluation suggestions.)

ANATOMICAL SITE
INNERVATION
METHOD OF EVALUATION
PROGRESSION OF FINDINGS
PROGRESSION OF SYMPTOMS
GROUP I
Tongue
XII
Inspect for fasciculations at rest
Fasciculations evident
Dysarthria (disturbance of lingual-alveolar consonants t, d, l, and so on)
Range of motion
Slow, incomplete lateral movementsLoss of lateral forceUnable to reach palate with mouth open
Inability to clear buccal sulcus of foodMarked dysarthria (slow rate and slurring of consonants)
Protrusion
Unable to protrude beyond lips
Oral transport difficultiesDietary changes
Perform rapid lateral motion
Unable to protrude beyond incisorsAtrophy evidentParalysis
Speech intelligibility problems
Lips
VII
Suck on gloved fingerSmile or curl lips over teethHold seal and blow out cheeks
Lack of suctionInability to complete a sealInability to purse lips
Inability to whistleInability to use a strawDysarthria (loss of bilabial consonants p and b)Drooling
GROUP 2
Palate
V, X, XI
Visual examination during phonation and stimulation of gagPuff out cheeks to check for nasal air leak (hold lips closed if necessary)
Unsustained or slow palatal elevationSoft palate fails to reach Passavant ridgeAbsence of palatal movement
Dysarthria (hypernasal speech)Inability to use a strawNasal air emission during speechNasopharyngeal reflex on swallowing
Muscles of mastication
V
Palpate during bite
Noticeable wasting
Chewing fatigue
Masseter, temporalis
Visual inspection for wasting
Unable to palpate contraction
Elimination of specific, tough foods from dietDietary changes (soft foods and liquids)Mouth breathing and drying of secretions
Pterygoids
Move jaw from side to side
No observable lateral jaw movement
Unable to use dentures
GROUP 3
Neck and shoulder
XI
Trapezius
Hold arm in coronal plane, hand externally rotated, as patient elevates arm against resistance while the trapezius is palpated
Progressive inability to raise the arm (often asymmetrical weakness)
Inability to comb hairInability to perform facial grooming
Sternocleidomastoid or mounted head support
Turn the head against resistance applied to opposite side of patient’s chin
Progressive weakness in turning the head against resistance (often asymmetrical)
Inability to lift head when supineInability to support head while sitting; wears neck collar, has weakness
Vocal cords
X
Mirror or fiberoptic laryngoscopy
Progressive loss of abduction of vocal cords: mild abductor weakness, near-midline paralysisParadoxical vocal cord movement
Strained or strangled voiceShort of breath (stridor usually not present because of impaired respiratory function)
GROUP 4
Extraocular muscles
III, IV, VI
Assessment of extraocular movements
Limitation of extraocular movement
Limitation of gaze
Respiratory group
Diaphragm
C3-5
Pulmonary function test or handheld respirometer for vital capacity
Diminishing vital capacity: 1.5-2.0 L
Shortness of breath during exertion if patient has remained active
Intercostal
C7-L3
Accessory muscles of respiration
VII, XI, XII, C5-8
CoughSustain a vowelBlow against a tissue
1.0-1.5 L
Weak coughChange in speech phrasing (5-10 syllables per breath)
0.5-1.0 L
Speech produced in syllable-by-syllable fashion (if vocal)Shortness of breath on swallowing
Goals of therapeutic intervention
Therapeutic considerations
Disuse atrophy.
Exercise or overwork damage.

AUTHOR
STUDY POPULATION AND SAMPLE SIZE
DURATION OF TRAINING
TRAINING MODALITY
TRAINING PROTOCOL
RESPONSE(S)
Vignos and Watkins, 1966291
Various neuromuscular diseases (NMDs) (24)
12 months
Weight training (multiple muscle groups)
Unspecified, but based on 10-repetition maximum (RM)
Strength increased; percentage increase correlated with initial strength
Milner-Brown and Miller, 198888
Various NMDs (12)
>12 months (variable)
Weight training (elbow flexion and knee extension)
Initially one set of 10 reps based on 15 RM performed on alternate days; gradually increased to a maximum of five sets 4 days/week; protocol individualized
Strength increased significantly when the initial degree of strength loss was not severe (<10%)
McCartney et al, 1988297
Various NMDs (12)
9 weeks
Weight training (arm curl and leg press)
3 days/week; initially two sets of 10-12 reps at 40% of 1 RM; gradually progressed to three sets of 10-12 reps (one set at 50%, 60%, and 70% of 1 RM); contralateral arm control
Strength and muscular endurance increased; considerable intersubject variability
Aitkens et al, 199389
Slowly progressive NMD (27) and able-bodied controls (14)
12 weeks
Weight training (elbow flexion, knee extension, grip one side only)
3 days/week; resistance at 30% of 1 RM; work increased commensurate with ability
Significant improvement in most isokinetic strength measures (not grip) in both groups; cross-training effect
Kilmer et al, 199490
Slowly progressive NMD (10) and able-bodied controls (6)
12 weeks
Weight training (elbow flexion, knee extension, one side only)
3-4 days/week; high-resistance exercise (resistance based on 12 RM; progressed from one to five sets of 10 reps)
Results mixed; increase in leg strength but decrease in arm strength in NMD
Lindeman et al, 1995325
MD (33) and HMSN (29); nonexercise control group
24 weeks
Weight training (knee extension and flexion, hip extension and flexion)
3 days/week; initially three sets of 25 reps at 60% of 1 RM; progressed to three sets of 10 reps at 80% of 1 RM
In MD group, no change in strengthIn HMSN group, increased strength of knee extensors; no adverse effects
Drory et al, 200193
ALS (25): randomly assigned to treatment or control groups
24 weeks
Moderate load, trunk and limbs, concentric contractions
Twice daily, 15 min per session
Treatment group: maintenance of strength, less fatigue, less spasticity, less pain, higher function
Aksu et al, 200292
ALS (26): convenience assignment to treatment and control groups
8 weeks
Breathing exercises, PNF, stretching, vs stretching and ROM and breathing exercises
3 days/week supervised vs home
Increased ROM, strength in treatment group; function sustained better in treatment group
Dawes et al, 2006326
Various NMDs: 11 randomly allocated to control group, nine to treatment group
8 weeks
Walking and strengthening exercises
Walking for 20 min at light to moderate intensity alternating days with progressive resistance and repetitions in strength
Treatment group had increase in leg muscle strength; no change either group in 2-min walk test
Dal Bello-Haas et al., 200794
ALS (27 early stage): randomly allocated to resistance exercises plus stretch or just stretch groups
24 weeks
PT-prescribed resistance exercises performed at home to patient tolerance
3 days/week resisted exercise plus stretch vs just stretch group
Slowed decline in treatment group and small strength gains Related posts:
![]() Multiple sclerosis
Multiple sclerosis
![]() Contemporary issues and theories of motor control, motor learning, and neuroplasticity
Contemporary issues and theories of motor control, motor learning, and neuroplasticity
![]() Electrophysiological testing and electrical stimulation in neurological rehabilitation
Electrophysiological testing and electrical stimulation in neurological rehabilitation
![]() Orthotics: evaluation, intervention, and prescription
Orthotics: evaluation, intervention, and prescription
![]() Movement dysfunction associated with cerebellar damage
Movement dysfunction associated with cerebellar damage
![]() Neonates and parents: neurodevelopmental perspectives in the neonatal intensive care unit and follow-up
Neonates and parents: neurodevelopmental perspectives in the neonatal intensive care unit and follow-up
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree