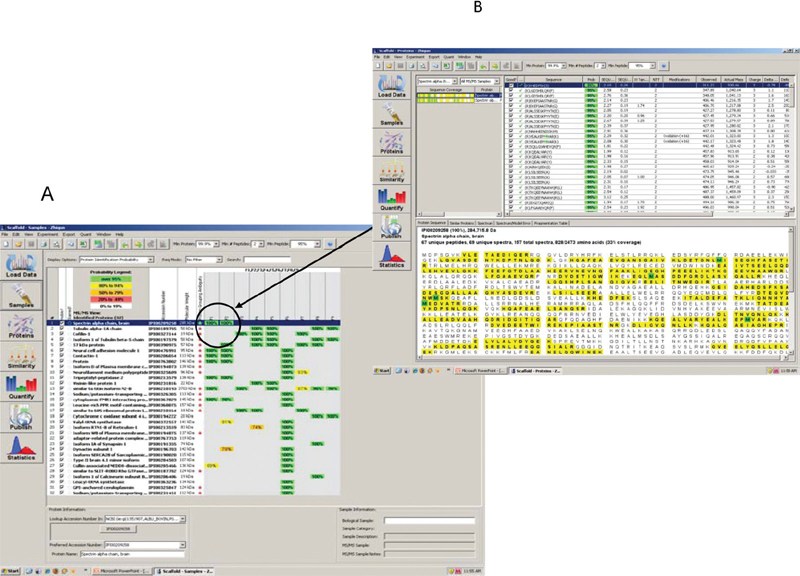
53 Key Points 1. The fields of neurogenomics/neuroproteomics are still in the developing stage due to several factors. The full potential of these areas remains to be explored and will reveal integral signature molecules known as biomarkers that can be associated with injury severity, cellular mechanisms, and biological networks specific to the particular brain disorder in question. 2. The complexity of the central nervous system (CNS) and associated brain disorders necessitates the use and development of relevant animal models of experimental CNS injury that can mimic human spinal or brain injury. 3. Introducing various advanced analytical tools (multidimensional separation techniques, neuroproteomics, neurogenomics, and biochemical testing) would facilitate the identification of key biological hits (proteins and/or genes) that can highlight the underlying mechanisms involved in the area of neural injury. Traumatic brain injury (TBI) is defined as neurotrauma caused by a mechanical force that is applied to the head.1 Annually in the United States, there are ∼ 2 million incidents that involve TBI. Of these, nearly 100,000 patients die, another 500,000 are hospitalized, and thousands of others suffer short- and long-term effects.2 The medical costs associated with caring for TBI patients are extremely high, as are the costs of lost productivity from TBI victims. Although this is one of the leading health epidemics in the country, there are currently no US Food and Drug Administration–approved treatments. Spinal cord injury (SCI) is considered among the most frequent causes of mortality and morbidity in every medical care system around the world. The incidence of SCI in the United States alone is estimated to be 11,000 new cases each year, affecting a total of 183,000 to 230,000 individuals.3 There are ∼ 900 to 1000 cases per million in the general population.4 TBI and SCI often result in permanent neurological damage due to the fact that neurons lack regenerative ability. After the initial injury occurs, many destructive processes cause secondary injury, such as edema and intracranial hemorrhages, resulting in more cell death and spreading tissue loss. Many studies have sought to find a way to enhance neuroprotective devices and find a mechanism that allows for neuronal cell regeneration.5,6 Studies have demonstrated the benefits of mild to moderate hypothermia in animal models of TBI and SCI.7 Stem cell–based treatment has been applied to experimental SCI models and has shown promising results.8 Genomics and proteomics could be very helpful in identifying specific proteins that are involved in such regeneration for the benefit of patients of TBI and SCI. Genomics and proteomics are powerful, complementary tools that play an important role in the study of neural injury. Over the past few years advances in the fields of neuroproteomics and neurogenomics have led to the discovery of many candidate biomarkers to help identify the mechanism of TBI. Identification of these biomarkers could lead to a more cost-effective and more time-efficient way to diagnose brain injury than the current techniques, such as magnetic resonance imaging (MRI) and computed tomographic (CT) scans. The expanding field of neuroproteomics gives a broader view of protein dynamics in TBI and SCI. Several studies have demonstrated the role of proteomics1,9 and genomics10,11 in providing significant insight into changes, modifications, and functions in certain proteins post-TBI. Proteomic approaches have been applied to studying rat models to identify novel proteins following SCI.11,12 A study by Kunz and colleagues used proteomics methods to identify novel proteins associated with chronic pain in SCI.13 Understanding the mechanism behind cell regeneration after SCI has been the aim of several studies.9,14 Genomics studies using microarray technologies include genome-based expression profiling in SCI rat models.15 Different neuroproteomic methods and protocols have been described in a book coedited by Ottens and Wang,16 including a two-dimensional (2-D) gel electrophoresis-based proteomic approach to study the proteome and phosphoproteome of the rat spinal cord lesion epicenter at 24 hours after spinal cord contusion.17 SCI elicits complex sequelae of destructive and neuroprotective cellular cascades. The alteration in transcription of the molecular events occurring during the post-SCI period holds the key for these cascades. Recent technological developments provide the ability to analyze changes in gene expression across tens to thousands of transcripts on the same tissue sample. Thus it provides a unique opportunity to take gene expression pattern profiles out of the specific SCI system under study and relate them to general CNS neuronal injury. There are several different technologies used for the simultaneous study of multiplex gene expression, including DNA microarrays, serial analysis of gene expression, Northern blot, nuclease protection assay (NPA), subtractive hybridization, and real-time reverse transcriptase-polymerase chain reaction (RT-PCR).18 Recently, mRNA differential profiles after SCI have been extensively performed to provide better understanding of the pathophysiology and formulation of a rational approach to treatment design after SCI.19 However, due to the different SCI experimental designs, such as SCI injury paradigm, animal strains, and differing magnitude, injury site, and data analysis, direct comparisons of results across different groups are yet to be fully appreciated when one is trying to generate a global picture of transcriptional regulation following SCI.20 The most common animal model used for SCI study is contusive SCI. Studies have examined the transcription changes over extended periods of time, ranging from hours to months after injury (Table 53.1). Considering the life span differences between rodents and human beings, we summarized the data into three categories: acute, subacute, and chronic phase post-SCI. Genes involved in inflammation, immune cell recruitment, and extravasation are highly upregulated during the acute phase postinjury. Transcriptional changes in inflammation-related molecules, such as cyclooxygenase 2 (COX2), levels of proinflammatory cytokines, such as interleukins (IL-1a, IL-1b, IL-4R, and IL-2Ra) and tumor necrosis factor (TNF)-α, are increased at early time points between 30 minutes and 6 hours post-SCI. This is followed by interstitial cell adhesion molecule (ICAM) and e- and p-selectin expression, which further recruit inflammatory cells to the injured sites, initiating secondary cytokine and inflammation signaling, such as IL-6, Fcg receptors (FcgRII and III) and classical complement (C1qb). Immediate early and cell cycle genes (cyclin D1, gadd45, krox24, NGF1-B); Scya2 or monocyte chemoattractant protein-1 (MCP-1); transcription factors, in particular those involved in cell damage and death, such as NF-κB, c-jun; suppressor of cytokine signaling 3 (SOCS-3), hsp70 and Bax, are upregulated in the epicenter samples post-SCI. Also, strong Janus kinase (JAK) and signal transducer and activator of transcription (STAT) activation might, however, represent an early attempt of the SCI repair and regeneration.20–22 Another common finding at early transcriptional changes after SCI is the down-regulation of ion channels and transport involved in cell excitability, such as N-methyl-D-aspartate (NMDA) receptor, glutamate transporter, and potassium ion channel, which are followed by cytoskeletal protein loss, such as neurofilament L and H (NFL, NFH), tau, and microtubule associated protein 2 (MAP-2). The spectrum of these gene expression changes reflects the initial tissue loss after SCI.23 After 24 hours postinjury, growth-associated molecules and growth factors start to be expressed, showing an attempt of the lesioned central nervous system (CNS) to regenerate and regrow. At 24 hours, a number of proteins have been shown to be upregulated, including nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), platelet-derived growth factor (PDGF), bone-morphogentic proteins (BMP), motor neuron-survival factor and fibroblast-growth-factor (FGF) receptor 1, insulin-like growth factor (IGF-I and II) and receptor, molecules possibly invovled in neuritogenesis (dynamin and attractin), and vascular adhesion molecules (VCAM).24 During the subacute period postinjury, the overall pattern of gene expression changes dramatically from damage-dominant transcriptional profile to one of active repair involving cellular proliferation and migration, which includes cell cycle genes (cyclin A2, cyclin G1, cell division cycle G25, oncogenes) and associated cytoskeletal and signal transcripts. These growth-promoting transcripts, such as cmyc, v-jun, and c-fos, are initiators of cell-cycle activity and protein synthesis and are followed at day 3 by large increase in transcripts coding for cellular division and the translational apparatus. A subset of growth factor gene families includes IGF, activin, follostatin, thyroid and steroid hormones, transforming growth factor b (TGFb), and those that moderate downstream growth factor signaling are substantially increased following SCI. Downregulation of neurotransmitter receptor, transporter, and synaptic molecules continues, such as NMDA receptor (NMDAR), glutamate receptor, rab3, and SNAP-25A, and contributes to secondary tissue damage.22,24–26 Several studies have investigated gene expression changes occurring in the chronic phase following SCI. The gene families expressed after 7 days postinjury are devoted to tissue repair and partial restoration of neural injury. Two categories are used according to the functional states of molecules after spinal injury. One group of genes is upregulated at chronic phase postinjury. These genes signify repair-directed transcript expression, which include matrix and blood vessel remodeling, antioxidant action, and blood–spinal cord barrier reestablishment. Gene families devoted to the regulation of angiogenesis and formation of tight junctions, including ICAM, P-selectin, VCAM-1, integrin ß-4, glypican-3, vitronectin, F-spondin, heparin sulfate proteoglycan core protein, tenasinx, Apo-E, and angiopoietin-2, are upregulated following SCI. Other transcripts, such as angiopoietin and pleiotrophin, IGF, BMP4 and 6 are also detected upregulated.22,27 Glutathione S-transferase, a known blood–spinal cord barrier marker, and glial fibrillary acidic protein (GFAP) continue to increase as late as 42 days after injury, suggesting blood–spinal cord barrier reconstruction. The genes for the reactive oxygen species (ROS) scavengers metallothionein 1 and II (MTI and II) and genes associated with pathways that induce MTI and II were robustly upregulated in the epicenter samples at 3 hours, 7 days, and 35 days.28,29 Structural protein vimentin, heme metabolism HO-1/HSP32, chaperon HSP27, transcriptor silencer factor-B, EGR1 (Krox-24), and osteopontin were observed to be upregulated in the acute phase and at 42 days.30 Expression of cathepsins B, C, D, K, and L was reported upregulated in the epicenter at 35 days and even longer at 42 days.30,31 In contrast, it has been reported that cathepsin B and D are involved in the degradation of serum amyloid A, which causes amyloid plaque in Alzheimer disease. The long-term increased expression of genes coding for cathepsin proteases may coordinate with modulation of the neurodegeneration.32 The upregulation of transcripts associated with neurodegeneration, such as synucleins, prion protein, amyloid precursor protein binding protein-1, and neurodegeneration-associated protein-1, at 30 to 90 days postinjury strongly supports the neurodegeneration process during the chronic period after SCI.22,32 Another group of altered genes during the chronic phase belongs to a family that decreases in expression immediately after injury and slowly returns to normal transcript levels at a later time. This group of genes includes those encoding neurotransmitter receptors (NMDAR, glutamate receptor), transporters [glycine transporter 1, gamma aminobutyric acid (GABA) transport proteins], ion pumps (Ca2+-ATPase, Na+/K+-ATPase), and synaptic proteins (synapsin I and II, syntaxin 2, synaptobrevin 2, synaptotagmin, synaptophysin, and SNAP25A).31,32 Rather than targeting transcription level, recent technology provides a powerful tool (microRNA array) to reveal the consequences after SCI at the gene regulation level.33 MicroRNAs (miRNAs) are a recently discovered class of small RNA molecules implicated in a wide range of diverse gene regulation mechanisms. Liu et al. identified three groups of miRNA that correlated the temporal expression postcontusion SCI.34 In summary, the development of technology of genes on the chips, it provides tremendous information and unravels the mechanisms underlying SCI to limit secondary damage and promote regeneration and ultimately improve the functional outcome of patients. Mass spectrometry (MS)-based proteomics workflow applicable to any type of biological sample, whether collected from human, animal, or cell culture models, can be organized into three stages: protein separation, protein identification and quantification, and bioinformatics analysis. Recent research articles describing the use of MS-based proteomics for studying protein expression changes in SCI have been published.11,12,14,35 Two-dimensional polyacrylamide gel electrophoresis (2-D-PAGE) has been the classical method of separation, visualization, and analysis of complex mixtures containing thousands of proteins. The first dimension involves isoelectric focusing, which separates proteins by their isoelectric point (pI) on a pH gradient strip. The second dimension resolves the proteins according to molecular weight, using a polyacrylamide gel. The spots are commonly visualized by Coomasie or silver staining methods. Despite the excellent resolving power of 2-D-PAGE, several limitations are present, including low sensitivity and poor reproducibility and the inability to resolve membrane proteins. This is due to their low abundance, high pI, and insolubility in aqueous buffers used in the isoelectric focusing step. Another separation technique relies on the separation by hydrophobicity or by charge where reproducible fractionation and eventually concentration of proteins of interest from the complex protein mixture is achieved. This method is referred to as cation anion exchange/polyacrylamide gel electrophoresis (CAX-PAGE), which is an approach developed in our laboratory that couples ion exchange chromatography for protein fractionation and 1D gel electrophoresis for protein separation, which is followed by MS analysis, as is discussed next.36–38 MS relies on the digestion of gel-separated proteins to peptides by sequence-specific proteases, such as trypsin. Tandem MS coupled to liquid chromatography (LC-MSMS) is among the major MS techniques used for proteomics analysis. The major advantage of tandem MS over other proteomics techniques is that it provides amino acid sequence information for the peptides that is more specific for identification of the protein. Tandem MS data are searched against a protein database using bioinformatics software. The peptides are matched to theoretical peptides in the database, and scores of how well the peptides match are generated. The database search output provides a list of identified proteins associated with the peptides. Figure 53.1 shows an example of a database search report of an excised band from a TBI sample. This illustrates that a single gel band can contain multiple proteins even after protein separation using a multidimensional approach like CAX-PAGE. Correct identification of the proteins depends on correct peptide identification. Another proteomics approach is quantitative proteomics. Quantitative proteomics provides an accurate picture of the dynamics of protein. Different strategies for quantifying proteins use sample labeling with stable isotopes as well as label-free methods. Label-free quantitative proteomics is an approach of growing interest among researchers. The stable isotope labeling method has been the gold standard in quantitative proteomics. It creates a mass shift to distinguish identical peptides from different samples with a single analysis. Alternative ways of introducing the stable isotopes into the peptides or proteins have been developed. One major labeling approach involves introduction of metabolic precursors with isotope labels in growth media of living cells. The multiplexing introduction of these proteomics techniques in assessing biomarker identification is an ideal approach for high throughput quantitative analysis of complex samples. Fig. 53.1 An example of the bioinformatic output of processed tandem mass spectrometry (MSMS) spectra collected from tryptic digests of excised gel bands. (A) At the set criteria (protein probability > 99.9% and peptide probability > 95%), several proteins were identified in each sample (band). (B) Peptide tandem mass spectra indicated presence of α-spectrin in sample F1.
Neurogenomic and Neuroproteomic Approaches to Studying Neural Injury
 Gene Expression Profiling of Experimental Traumatic Spinal Cord Injury
Gene Expression Profiling of Experimental Traumatic Spinal Cord Injury
 Mass Spectrometry–Based Proteomics
Mass Spectrometry–Based Proteomics
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


