Chapter 19 Immune Tolerance Defects in Lupus
Lack of central tolerance by negative selection in the thymus or bone marrow is an initial step in the development of autoreactive T or B cells, respectively.1 Negative selection to ubiquitous self-antigens occurs in the cortex of the thymus, whereas negative selection to tissue-restricted self-antigens that are targeted in organ-specific autoimmune diseases occurs in the medulla of the thymus.2,3 The loss of central T-cell tolerance, however, might be of little pathologic consequence, as robust peripheral tolerance mechanisms provide major control against pathologic autoimmunity.2,4 Similarly, B cells that encounter self-antigens in the periphery face several tolerance checkpoints.5,6 Patients with SLE have defects in B-cell tolerance at several of these checkpoints, including one at the transition from the early immature to the immature stage, another at the transitional to mature stage, a germinal center (GC) entry checkpoint, and a checkpoint between naïve and antigen-experienced B cells.7,8 Belimumab, the only drug approved by the U.S. Food and Drug Administration (FDA) for SLE in more than 50 years, is assumed to act at one of these peripheral B-cell tolerance checkpoints via deletion of autoreactive transitional and naïve B cells.8,9 It remains to be determined, however, whether treatment with belimumab also depletes a subset of transitional and naïve B cells that act as regulatory B (Breg) cells.10–12 Depletion of such protective B cells can potentially tamper the therapeutic efficacy of belimumab.
Immune Tolerance
Lymphocyte Homeostasis and Immune Tolerance
The immune system is unique in its ability to maintain a state of equilibrium despite its continuous exposure to self-antigens as well as its requirement to mount an adequate response to a variety of foreign antigens. After responding to an antigen, the immune system returns to its original state, so that the numbers and functional status of lymphocytes are reset at roughly the original state. This process, known as lymphoid homeostasis, allows the immune system to respond to new antigenic challenges. The size and content of the preimmune lymphocyte repertoire are tightly regulated, as new emigrants from the lymphoid organs compete for “space” with resident cells.13 Several groups have tried to define factors that control naïve and memory T-cell homeostasis under lymphoproliferative or lymphopenic conditions.14 There has been a renewed interest in the hypothesis that in lymphopenic conditions, T cells expand to reestablish homeostasis by a process dependent on self–major histocompatibility complex (MHC)–peptide recognition and on the availability of cytokines that can promote the proliferation and survival of lymphocytes. Such lymphocyte expansion is believed to be a normal physiologic process. The constant recurrence of this process, however, might lead to the selection and accumulation of high-affinity self-reactive T-cell clones and ensuing autoimmune disease.15 Experimental support for this hypothesis was reported in a study that showed that autoimmune nonobese diabetic (NOD) mice have reduced numbers of CD4+ T and B cells in comparison with control mouse strains.16 Increasing T-cell numbers, such as by immunization with complete Freund’s adjuvant (CFA), increases B-cell numbers as well and protects these mice from autoimmune diabetes. Interestingly, self-reactive T-cell receptor (TCR) transgenic T cells expand in the lymphopenic NOD mice, but not in NOD mice “filled” (reconstituted) with syngeneic T cells, in CFA-immunized NOD mice, and in congenic B6.idd3.NOD mice that have normal T- and B-cell numbers. Thus, lymphopenia and the resulting compensatory homeostatic expansion of effector lymphocytes reactive with self-antigens may precipitate autoimmunity.16 Another example of lymphopenia-induced autoimmunity in rodents is the development of autoimmunity after neonatal thymectomy, discontinuation of cyclosporine treatment, or total lymphoid irradiation. Lymphopenia also accompanies human autoimmune diseases, such as SLE and Sjögren syndrome.17
Lymphocytes with receptors specific for self-antigens are generated continuously in the body, yet most individuals maintain a state of unresponsiveness to their own antigens, a process referred to as self–immune tolerance. Thus, immune tolerance can be broadly defined as a physiologic state in which the immune system does not react harmfully against the components of an organism that harbors it or against antigens that are introduced to it.18 Harmful responses are prevented by a variety of mechanisms that operate during development of the immune system and during the generation of each immune response. These mechanisms can be broadly classified into four major groups: Central tolerance—which implies induction of tolerance in developing lymphocytes when they encounter self-antigens in the thymus or bone marrow—ensures tolerance to self-antigens that are present in high concentrations in the bone marrow and thymus. This process occurs by induction of apoptosis of self-reactive lymphocytes also known as clonal deletion. Peripheral tolerance is maintained by mechanisms that operate on mature lymphocytes once they exit the primary lymphoid organs. Ignorance may be the mechanism of tolerance for these self-antigens, which is believed to operate when the self-antigen is sequestered in anatomic sites, which are inaccessible to lymphocytes. Clonal anergy is another mechanism of lymphocyte tolerance in which the lymphocyte is functionally unresponsive following antigen encounter but remains alive for extended periods in a hyporesponsive state.19 Self-antigen recognition without co-stimulatory signals is widely believed to induce lymphocyte anergy. However, the conditions or factors that determine whether a self-antigen can be functionally ignored or induces anergy remain to be fully understood. More importantly, so far we do not know which self-antigens can induce which form of self-tolerance or what is the relative contribution of each of these mechanisms in shaping the normal immune repertoire. There is also no proper understanding of which characteristics of a self-antigen can lead it to undergo central tolerance, peripheral tolerance, clonal ignorance, or clonal anergy. Nevertheless, substantial progress has been made in unraveling these basic tolerance mechanisms that are common to both B and T lymphocytes. Because current knowledge supports loss of tolerance in both B and T cells in eliciting pathologic autoimmunity, we discuss them separately (Table 19-1).
TABLE 19-1 Mechanisms of Self-Tolerance in T and B Cells
| MECHANISM | PRESENT IN T CELLS? | PRESENT IN B CELLS? |
|---|---|---|
| Clonal deletion | Yes | Yes |
| Ignorance | Yes | Yes |
| Anergy | Yes | Yes |
| Immune deviation | Yes | No |
| Regulatory T/B cells | Yes | Yes |
| Receptor editing | No | Yes |
Mechanisms Underlying T-Cell Tolerance
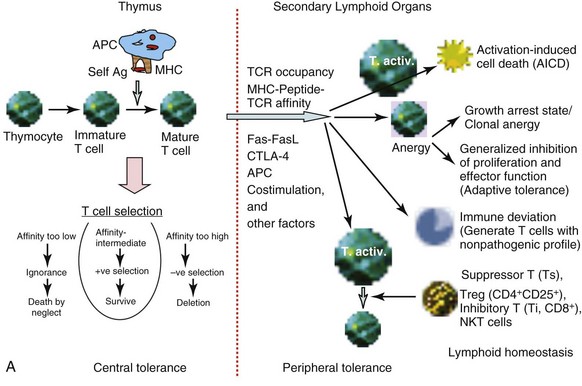
Tolerance of self-reactive T cells occurs in both a central tolerance mode, occurring in the thymus, and in peripheral tolerance mode, occurring in the peripheral lymphoid organs. The sites of tolerance and potential mechanisms are depicted in Figure 19-1, A.
Thymic Selection
The recognition of self-peptides, in association with self-MHC molecules, presented to differentiating T cells by antigen-presenting cells (APCs) present in the thymus results in thymic selection of T cells.1 This thymic selection process ensures that mature T cells are both self-MHC–restricted and self-tolerant. When the TCRs on a pre–T cell thymocyte are engaged, the thymocyte can be either positively or negatively selected, depending on the balancing effects of several other factors regulating this process. Thymic selection begins at the double-positive (DP; CD4+CD8+) stage in the thymus (when the α and β chain genes are expressed) and beyond. This process has two important outcomes: MHC restriction (positive selection) and central tolerance (negative selection). Thymic cortical epithelial cells function as the effector cells in a process known as positive selection. In positive selection, T cells that bear a TCR that can bind self-MHC are selected to survive and proliferate. T cells that are not positively selected are triggered to undergo apoptosis. Positively selected thymocytes must go through a second phase of selection known as negative selection. During negative selection, any T cell that is presented with antigenic peptide bound to MHC within the thymus is triggered to undergo apoptosis if the avidity between the TCR and the MHC/self-peptide is too strong.
The self-peptides encountered in the thymus are derived from proteins expressed in the thymus as well as other proteins brought to the thymus via the bloodstream. Moreover, medullary thymic epithelial cells (mTECs) express the autoimmune regulator (Aire) transcription factor that allows promiscuous gene expression.20 Aire allows mTECs to express not only antigens that are ubiquitously expressed but also antigens that are exclusively restricted in organ-specific cells. The Aire-controlled expression of tissue-restricted self-antigens provides a dynamic mechanism by which organ-specific self-tolerance is achieved. Mutations in the gene encoding Aire have been recognized as the molecular cause of the autoimmune polyendocrine syndrome type 1 (APS-1). In addition, dendritic cells (DCs) can functionally remove autoreactive T cells and express tissue-restricted self-antigens released from mTECs.21,22 Surviving T cells continue to migrate to the medulla, where they undergo full maturation and finally leave the thymus through the postcapillary venules or efferent lymphatics. Data now suggest that negative selection to ubiquitous self-antigens occurs in the cortex of thymus without medullary involvement, whereas positive selection and migration to the medulla are required for negative selection to tissue-restricted self-antigens.2,3
Although thymic selection should enable the deletion of all self-reactive T cells, this process is not perfect because not all peptides that an organism may encounter in the lifetime are presented in the thymus. Other variables, such as peptide concentrations, affinity of TCRs, and state of APCs in the thymus, may all determine whether the threshold for receptor occupancy is reached for the positive or negative selection to occur. Potentially self-reactive T cells that escape central tolerance can still be tamed through several backup mechanisms for maintenance of self-tolerance.2,4 These peripheral tolerance mechanisms include antigen-specific unresponsiveness or anergy, immune deviation, and elimination after repeated activation.23 Variables that determine whether peripheral deletion proceeds efficiently include extent of TCR occupancy, affinity of antigenic peptide for the MHC, and affinity of TCR for the antigen peptide complex. High antigenic dose and chronic stimulation favor elimination both in CD4+ and CD8+ T cells. The silencing of T cells upon persistent activation in the periphery may thus represent a continuous process, ranging from the activation to unresponsiveness to deletion, with T-cell signal strength and exposure time together determining the outcome. A major mechanism for peripheral deletion of activated T cells involves activation-induced cell death (AICD) via the Fas-FasL pathway, as suggested by studies in mouse models, in which mutations in these molecules are associated with the development of autoimmunity. In mice deficient in Fas (MRL/Faslpr/lpr [MRL/lpr]) or the Fas ligand (FasL) (gld mice), severe lymphoproliferative autoimmune disease develops as a result of accumulation of activated T cells. Mutations in Fas are associated with autoimmune disease in humans as well.24 Some mouse strains carrying gld or lpr mutations demonstrate inflammatory disease, whereas the same mutations on other genetic backgrounds cause only excessive lymphoproliferation.25 In humans, not all subjects carrying mutations in Fas or FasL experience autoimmune disease.24,26 Thus, additional mechanisms must contribute to peripheral tolerance of autoreactive T cells.
Inhibitory co-stimulatory molecules like cytotoxic T-lymphocyte antigen 4 (CTLA-4) and programmed death 1 (PD-1) have been implicated in peripheral tolerance. These two molecules play distinct regulatory roles. Although CTLA-4 is involved in regulating initiation of immune response in the lymph node, PD-1 pathways act late at the tissue site to limit T-cell activation.27
Induction of Anergy
Anergy induction is another mechanism of lymphocyte tolerance, in which the lymphocyte is functionally unresponsive after antigen encounter but remains alive for extended periods in a hyporesponsive state.19 The basic types of T-cell anergy fall in two groups. One is the principally growth arrest state clonal anergy, and the other is adaptive tolerance or in vivo anergy, a generalized inhibition of proliferation and effector functions.28 According to the two-signal model of T-cell activation versus anergy, the APCs having the ability to offer T cells the prerequisite triggering of TCRs (signal 1) and co-stimulation (CD28/B7; signal 2) induce T-cell activation. However, not all APCs have the ability to offer T cells both of these signals, and signaling through the TCR alone induces a state of functional unresponsiveness, or clonal anergy. This could happen via two pathways: One is the direct inhibition of CD28 signaling by “anergy factors,” and the other involves an indirect effect on cell cycle progression through growth factors such as interleukin-1 (IL-2).29,30 Some studies have led to a better understanding of the cell-intrinsic program that establishes T-cell anergy. During the induction phase of anergy, “incomplete” stimulation of T cells (TCR triggering without co-stimulation) leads via calcium influx to an altered gene expression program that includes upregulation of several E3 ubiquitin ligases. When the anergic T cells contact APCs, intracellular signaling proteins are monoubiquitinated and targeted for lysosomal degradation, thus decreasing intracellular signaling and also resulting in decreased stability of the T cell–APC contact.31 Ubiquitin ligases that have been implicated in T-cell anergy are c-Cbl, Cbl-b, GRAIL, ITCH, and Nedd4.32 Interplay of these ubiquitin ligases has been shown to regulate T-cell anergy.
Immune Deviation
The immune system has also evolved to have a functional mechanism of tolerance in the face of persistent T-cell activation. Skewing of a T-cell response into a lineage that does not mediate disease and that prevents development of harmful T-cell responses is called immune deviation. In NOD mice, in which diabetes spontaneously develops, the presence of T-helper 1 (Th1) cells in islets was found to be associated with the clinical disease, whereas resistance to disease is associated with predominance of cells producing Th2-like cytokines.33 Similarly, in experimental autoimmune encephalomyelitis (EAE) models, the Th1 responses are generally pathogenic, whereas Th2 responses are protective. Paradoxically, the blockade of Th1 differentiation in IL-12 receptor 2–deficient mice results in more severe EAE.34 This led to the discovery of another T-helper cell type, known as Th17 cells because of their production of the proinflammatory cytokine IL-17.35 It was quickly recognized that Th17 cells mediate major pathogenic functions in many autoimmune diseases, such as multiple sclerosis (MS), rheumatoid arthritis (RA), inflammatory bowel disease (IBD), diabetes, Sjögren syndrome, and psoriasis, and even Th2-mediated inflammatory diseases such as asthma.36 Specific mechanisms, which allow skewing of T-cell immune deviation into Th1, Th2, Th17, and T-regulatory (Treg) cells, are not fully understood. Several explanations for the apparent dichotomy have been proposed, including the role of the types of APCs participating in the immune response, modulation of the co-stimulatory molecules, cytokine secretion, and signal transduction pathways. Interestingly, both Th17 and Treg cells can develop from naïve CD4+ T-cell precursors under the influence of transforming growth factor β1 (TGF-β1), depending on the other cytokines in the milieu. Therefore autoimmunity may result when the differentiation of CD4+ T cells is favored toward differentiation of Th17 cells instead of Treg cells.
Regulatory, Suppressor, or Inhibitory T cells
A number of T-cell subsets have been shown to regulate or suppress autoimmunity. The most studied of these subsets, Tregs, which are identified as CD4+CD25+Foxp3+ cells, are described in another chapter (‘Regulatory Cells’). We have also described CD8+ inhibitory T cells that, by producing TGF-β, prevent or suppress autoimmunity,37 and CD8+ cytotoxic T cells, which can ablate autoreactive B cells.38 Natural killer T (NKT) cells, especially those that express invariant TCR, can induce self-tolerance in the periphery via multiple mechanisms, including the regulation of autoreactive B cells.39–42
Mechanism of B-Cell Tolerance
As with T cells, tolerance of self-reactive B cells occurs in both a central tolerance mode occurring in the bone marrow and in peripheral tolerance mode occurring at different stages of maturation of B cells as well as at the level of mature B cells, as depicted in Figure 19-2A.
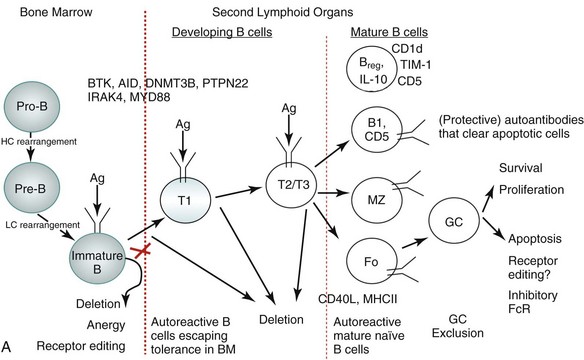
More than half of all newly generated immature B cells in the bone marrow of healthy individuals appear to be polyautoreactive and capable of binding self-antigen, including nuclear antigens.43,44 Elaborate control mechanisms must therefore exist to remove such potentially autoreactive B cells, ensuring self-tolerance. In fact, extensive studies in mouse models and some in humans with regard to B-cell selection suggest that there are a number of distinct tolerance checkpoints during B-cell development and maturation,43,45 which can be broadly categorized into three stages. First, there is an initial checkpoint during the maturation of B cells in the bone marrow; second, there are many checkpoints during B-cell development in the periphery; and finally, there are checkpoints involving mature B-cell subsets (see Figure 19-2A).
The majority of polyreactive and antinuclear antibody B cells are removed at the immature B-cell stage in the bone marrow,43 which essentially involves three mechanisms: deletion, anergy, and receptor editing.5,44,46,47 B-cell receptor (BCR) signaling strength and the physical nature of the self-antigen (soluble versus membrane-bound) play major regulatory roles in the selection process.5,48 Stronger signals result in apoptosis of B cells, called clonal deletion. Weaker signals render the B cell unresponsive to antigen stimulation, a state known as anergy. Anergic cells are susceptible to early death. In some B cells, re-expression of recombinant activating gene (RAG) proteins allows replacement of self-reactive receptors with non–self-reactive ones, a process known as receptor editing.
In the periphery, most remaining potentially autoreactive B cells are removed when newly emigrant B cells transition into naïve immunocompetent lymphocytes.43 To develop from the immature state in the bone marrow to the mature naïve state in the peripheral lymphoid organs, a B cell must survive several checkpoints.45 The first checkpoint is between the immature cell in the bone marrow and the transitional T1 cell in the spleen. The second is between the T1 and more mature T2/3 state, and the third is between the T2/T3 stage and mature B cells. This process depends not only on the strength of the BCR signal the B cells receive if and when they encounter a self-antigen but also on competition with non–self-reactive B cells for B cell–activating factor of the tumor necrosis factor family (BAFF).8,49 The transitional B cells can be rescued from negative selection by co-stimulatory signals. For example, CD40 engagement by CD40 ligand (CD40L) can rescue B cells destined to undergo BCR-mediated apoptosis. Apoptotic cells, a source of endogenous TLR ligands, can activate Toll-like receptors (TLRs), which also promote T-independent class switching and differentiation of B cells. Apoptotic cells are normally removed from circulation by macrophages, thus preventing any autoreactivity via this mechanism.
B cells that escape tolerance mature into immunocompetent B cells having the phenotype of one of the following B-cell subsets: B-1 cells, marginal zone (MZ) B cells, short-lived plasma cells, and GC-matured long-lived plasma cells. The B1 cells express CD5, are restricted in diversity, and fail to generate a memory population. Self-reactive B-1 cells bearing low-affinity BCRs normally home to the peritoneal cavity (in mice) and produce autoantibodies that are thought to help avoid pathogenic autoreactivity by clearing apoptotic cells.50 The human equivalent of B-1 cells, which express CD5, are normally present in the naïve repertoire, but they are usually excluded from the GC reactions.51 The MZ B cells that mature rapidly into plasmablasts can produce autoantibodies.52 How tolerance is regulated in the MZ B cell subset is unclear. Our group has recently shown that invariant NKT cells that normally activate MZ B cells control MZ B–cell homeostasis by promoting their activation-induced cell death and inhibiting their proliferation,41 and thus reducing autoantibody production.42 The follicular B cells, after they encounter antigen and receive T-cell help, generate GCs to mount an affinity-matured antibody response and generate memory B cells.
Germinal center exclusion of potentially autoreactive B cells is an important checkpoint in mature B cells to preclude class switching and somatic hypermutation. In GCs, a stringent balance of proliferative and apoptotic signals is required to prevent the survival of self-reactive B cells while ensuring expansion of the normal B-cell repertoire. Mechanisms of positive and negative selection at the level of GC are not well understood. Receptor editing might contribute to negative selection at this stage,53 and inhibitory Fc receptor FcγRIIB may regulate B-cell survival in the GC,54 whereas T cells may serve to mediate positive selection of GC B cells. Finally, autoreactive CD138+ preplasma cells can be prevented from differentiating into antibody-secreting plasma cells by long-term BCR engagement by self-antigen.
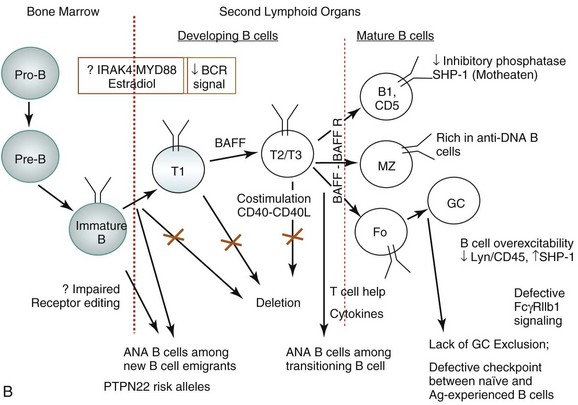
Studies of B-cell tolerance in patients with primary immune deficiency diseases can be instructive for tolerance breakdown in SLE.6 Indeed, risk alleles encoding variants altering BCR signaling, such as PTPN22 alleles associated with the development of SLE, interfere with the removal of developing autoreactive B cells. Patients with deficiencies of IRAK4, MYD88, and UNC-93B, which are involved in TLR signaling, also have a defective central B-cell tolerance. Defective central B-cell tolerance is also seen in patients with activation-induced cytidine deaminase (AID) deficiency and X-linked agammaglobulinemia who carry mutations in the BTK gene, which encodes an essential BCR signaling component. In contrast, CD40L− and MHC class II–deficient patients displayed only peripheral B-cell tolerance defects. Thus, central B-cell tolerance is mostly controlled by intrinsic B-cell factors that regulate BCR and TLR signaling, whereas peripheral B-cell tolerance seems to involve extrinsic B-cell factors, such as Treg cells and serum BAFF concentrations in humans. Furthermore, defects in peripheral B cell tolerance mechanisms are also detected in patients who have defects in central B-cell tolerance.
Studies have now demonstrated the existence of a subset of IL-10–producing splenic B cells that can serve as regulatory B cells. Bregs promote tolerance in a number of autoimmune models, including collagen-induced arthritis.10 Lacking specific markers, such Breg activity appears to reside within uncommon IL-10–expressing B cells scattered within various B-cell subpopulations. One study characterized Bregs as CD1dhiCD5+ B cells expressing T-cell Ig domain and mucin domain protein 1 (TIM-1) molecule.12
Immune Tolerance Defects in Lupus
Although substantial progress has been made in understanding fundamental mechanisms of self-tolerance, how impairment in this process causes autoimmune disease remains largely unclear. A number of mechanisms have been proposed, and a few have been demonstrated in animal models of autoimmunity. As summarized in Table 19-2, some of these mechanisms depend on alterations in autoantigen itself,55–57 some on changes in the processing and presentation of autoantigen at the level of APCs, some on changes in the T and B cells, and some on aberrant immune regulation. On the basis of our current understanding, alterations at different levels may account for loss of self-tolerance in different animal models and probably in different subsets of SLE, and multiple impairments could well account for the loss of self-tolerance in a single model or patient.55
TABLE 19-2 Mechanisms of Tolerance Breakdown in Lupus
| SITE OF ALTERATION | ALTERATIONS |
|---|---|
| Autoantigen | |
| Antigen-presenting cells (APCs) | |
| T cells | |
| B cells | |
| Regulatory T cells and cytokines |
Abnormalities at the Level of Autoantigens in Causing Tolerance Breakdown
Impaired removal of apoptotic cells could contribute to an overload of autoantigens, which can cause prolonged activation of immune cells. There are also several examples of autoimmunity being triggered by responses to foreign antigens via molecular mimicry. That only certain self-proteins frequently elicit an autoimmune response has intrigued many investigators to speculate that autoimmunity might occur because altered self or modified self serves as a potential source of autoantigen. Several mechanisms, including somatic mutations, genetic polymorphisms, alternative splicing, and posttranslational modifications, could generate epitopes for which the immune system is not tolerized.58 Defective apoptosis and altered antigen processing can also result in the generation of neoepitopes. The modified antigens can be taken up, processed, and presented by APCs and recognized by existing potentially self-reactive B and T cells, resulting in breakdown of tolerance and induction of autoimmunity. These mechanisms are described in Chapter 21 ‘Autoantigenesis and Antigen-based Therapy and Vaccination in SLE’.
Impairment of Antigen-Presenting Cell Function in Tolerance Breakdown
Using nucleosome-specific TCR transgenic mice, Michaels demonstrated that thymic DCs from lupus-prone mice are less efficient than those from normal mice in presenting naturally processed nucleosomal peptides in the steady state.4 Thus, insufficient presentation of self-antigens in the thymus may account for positive selection and/or lack of negative selection of autoreactive T cells in an autoimmune-prone background.
Specialized DC subsets that reside in peripheral tissues carry antigens from the tissue to draining lymph nodes to maintain tolerance to respective tissue antigens in steady state, thus avoiding autoimmunity. This process of peripheral tolerance is impaired in lupus, because Langerhans DCs in the skin of lupus-prone MRL/lpr mice display impaired capacity to migrate to draining lymph nodes in comparison with cells in normal strains.59 Thus, impaired capacity of DCs to present self-antigens in the thymus4 or reduced ability of tissue-resident DCs to migrate and carry self-antigens to draining lymph nodes59 may contribute to a breakdown in central or peripheral tolerance, respectively.
Molecular mechanisms underlying DC defects in SLE are not defined. One study demonstrated a critical role for B-lymphocyte-induced maturation protein 1 (BLIMP-1), a key regulator of plasma-cell differentiation in B cells and of effector/memory function in T cells, in the tolerogenic function of DCs.60 The investigators showed that a diminished expression of BLIMP-1 in DCs results in increased production of IL-6, preferential differentiation of follicular T-helper (TFH) cells, and development of a lupus-like serology in female but not male mice. Of particular relevance to human SLE, a polymorphism of BLIMP-1 is associated with SLE.
B cells may also serve as important APCs in breaking T-cell tolerance. Using anti–small nuclear ribonucleoprotein immunoglobulin (anti-snRNP Ig) transgenic mice, Yan and Mamula showed that whereas both normal and autoimmune (MRL/lpr) mice harbor autoreactive T cells, transgenic B cells can tolerize autoreactive T cells in the periphery of normal mice only.61 Thus, B cells (anti-snRNP transgenic B cells in this case) served as important APCs for T cell tolerance in normal mice and for T-cell activation in MRL mice. The study further suggested that anti-snRNP B-cell anergy in normal mice could be reversed by autoreactive T cells from autoimmune mice in a cognate manner, indicating an important role of T cells in the development of lupus (as described later).
T-Cell Abnormalities Contributing to Tolerance Breakdown
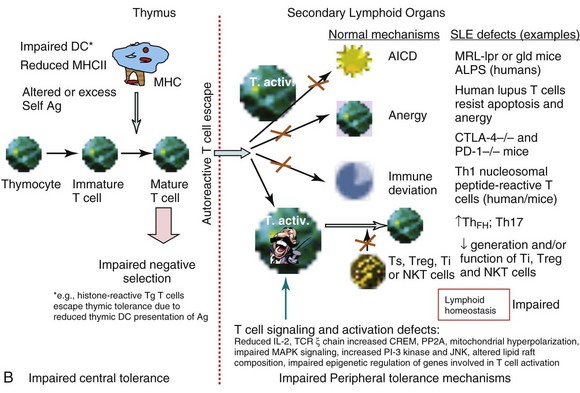
Studies of T-cell tolerance using animal models or human T cells have revealed a plethora of impairments at almost every level of central or peripheral tolerance mechanisms in SLE (see Figure 19-1, B).
Impaired Clonal Deletion of Lupus Autoreactive T-Helper Cells
To determine whether lupus T cells arise as a consequence of failed negative selection, Datta used transgenic mice expressing TCR of a pathogenic autoantibody-inducing Th cell that was specific for nucleosomes and its histone peptide H471-94. The investigators found that whereas thymocytes carrying lupus TCR were deleted in normal mice, such negative selection did not occur in the thymus of lupus-prone (SWR × NZB)F1 (SNF1) mice.4 Thus, impaired central tolerance may contribute to the positive selection of autoreactive pathogenic Th cells in lupus (see Figure 19-1, B). This idea is further supported by a study in which procainamide-hydroxylamine (PAHA), a drug that induces lupus in humans, has been found to interfere with central tolerance mechanisms in the thymus, resulting in the emergence of chromatin-reactive T cells followed by humoral autoimmunity in C57BL/6xDBA/2 F1 mice.62 To address this issue in humans, T cells from patients with SLE were cultured with thymic stromal cells. In these experiments, T cells from patients with SLE are more resistant to induction of apoptosis by thymic stromal cells than normal T cells. Thus, SLE T cells have intrinsically acquired a mechanism to evade central tolerance mechanisms in SLE, whereby interactions between thymic stromal and lymphoid cells lead to subsequent survival of autoimmune T cells.63
Neonatal and Adult Tolerance to Exogenously Administered Peptide Antigens in Lupus
To understand mechanisms and outcome of tolerance induction in lupus, our group administered MHC class II–binding foreign or self peptides, namely, hen egg lysozyme (HEL) 106-116 or self immunoglobulin A6.1 VH58-69, to newborn lupus (NZB/NZW F1) or normal (BALB/c) mice.64 A comparable level of tolerance, as measured by peptide-specific T-cell proliferation and IL-2 production in response to subsequent peptide challenge, was induced in both lupus-prone and normal mice. Lupus-prone mice, however, had increased anti-DNA antibody production in response to a neonatally administered self-VH peptide. Comparable levels of tolerance were also induced in adult lupus-prone and normal control mice, when peptide antigens were administered intravenously in high doses of soluble form or intraperitoneally in high doses of emulsified form.65–67 The older lupus-prone animals, however, tended to have relatively more leakiness in tolerance, particularly in Th functions and peptide-specific antibody responses (RR Singh, unpublished data, 1999). These studies demonstrate lack of a major tolerance defect in the induction of experimental tolerance in lupus-prone mice.
Intact Central Tolerance but Impaired Peripheral T-Cell Control Mechanisms
Several groups have studied mechanisms and outcome of tolerance induction in lupus using transgenic mice expressing TCR of a T cell specific for a conventional peptide antigen (e.g., pigeon cytochrome C [PCC]). In the pigeon cytochrome C peptide TCR transgenic model, the relevant antigen exposure results in intrathymic deletion of immature CD4+D8+ double-positive thymocytes, TCR downregulation, and thymocyte apoptosis, which are comparable in a nonautoimmune mouse strain (B10.BR) and an autoimmune-prone MRL/MpJ strain.68 Thus, central tolerance to a conventional antigen is intact in lupus-prone MRL mice. Using the NZB model, another study inferred that there is no generalized T-cell tolerance defect in lupus-prone mice.69 Thus, lupus-autoreactive T cells may arise in the setting of incomplete but qualitatively normal tolerance or as a result of defects in peripheral control mechanisms.
Using gene microarray profiling and functional and biochemical studies, one study showed that activated T cells of patients with SLE resist anergy and apoptosis (see Figure 19-1, B) by upregulating and sustaining cyclooxygenase-2 (COX-2) expression, along with the antiapoptotic or survival molecule c-FLIP (cellular homolog of viral FLICE inhibitory protein).70 Inhibition of COX-2 causes apoptosis of the anergy-resistant lupus T cells by augmenting Fas signaling and reducing c-FLIP. Studies with COX-2 inhibitors and Cox-2–deficient mice confirmed that anergy-resistant lupus T cells, and not cancer cells or other autoimmune T cells, selectively use this COX-2/FLIP antiapoptosis program. Thus, an imbalance in the proapoptotic and antiapoptotic mechanisms may contribute to the persistence of autoreactive clones.70
Studies in animals also show that CD4+ T cells from lupus mice are more resistant than those in nonautoimmune mice to anergy induction (see Figure 19-1, B). Anergy avoidance in the periphery may be one of the causes for abnormal T-cell activation in response to self-antigen in SLE.71 Indeed, T cells from patients with SLE and lupus-prone mice display phenotypes of in vivo activation, as defined by expression of CD25, HLA-DR, and CD40L.72,73 Additionally, increased expression of perforin and granzyme on CD8+ T cells correlates with disease activity in patients with SLE.74 Thus, T-cell activation appears to be a hallmark of disease development in SLE. However, the mechanisms that cause T cells to become hyperactivated or overexcitable have not been well defined, except for those described in the following section on T cell–signaling defects.
Studies in lupus mice show that heightened response to peptide antigens, particularly those with low affinity for TCR, appears to drive the polyclonal T-cell activation.75 Many studies have also demonstrated the presence of intrinsic T-cell abnormalities, such as diminished activation thresholds, in patients and mice with SLE. The following sections narrate efforts of several laboratories to define such intrinsic T-cell abnormalities in lupus.
T Cell–Signaling Defects in SLE
T cells use a cell surface multi-subunit structure, the TCR/CD3 complex, as an antigen-specific recognition site. The TCR α/β or γ/δ chains are the antigen-binding sites but, because of having very short cytoplasmic domains, they are not capable of any signal transduction, which is carried out by the CD3 complex. Human and murine SLE T cells, when stimulated through the TCR/CD3 complex, exhibit several abnormalities in T-cell signaling (see Figure 19-1, B). These include aberrant tyrosine phosphorylation, altered calcium flux, and heightened mitochondrial potential. A major and well-studied outcome of this aberrant signal transduction in SLE T cells is reduced IL-2 production, a phenotype of lupus T cells observed 30 years ago.76,77 Reduced response to IL-2 by T cells accompanied reduced IL-2 production in some patients with SLE.76 One study found a severe defect in IL-2 production by mononuclear cells from all 19 subjects, who were patients with SLE, regardless of the stimulant used and irrespective of the patients’ disease activity.78 Defective IL-2 production has also been reported in mouse models of lupus, including MRL/lpr, BXSB, and BWF1 mice.77,79 In MRL/lpr mice, reduced IL-2 production precedes the onset of clinical illness and becomes increasingly severe with age79 (S Dubey and RR Singh, unpublished data, 2005). Spleen cells from MRL/lpr mice also fail to respond normally to IL-2.79 It is therefore important to focus on the IL-2 defect in SLE T cells, because it acts as an essential regulator of immune response by promoting activation of the immune system and terminating it when required by inducing activation-induced cell death of autoreactive T cells. In fact, treatment of MRL/lpr mice with the Il-2 gene delivered via Vaccinia virus or attenuated Salmonella vectors results in significant improvement in lupus disease.80 Consistent with reduced IL-2 production, proliferative responses of T cells from patients with SLE, when the cells are cultured with thymic stromal cells, are lower than those of their normal counterparts.63 In addition to being a potent T-cell growth factor, IL-2 is essential for immune tolerance. Accordingly, mice deficient in IL-2 succumb to a rapidly progressing autoimmune disease that is caused by an uncontrolled activation of T and B cells.81 It was thereafter discovered that IL-2 was critically required for the development, homeostatic maintenance, and suppressive function of Treg cells.82 A number of reports have found a low prevalence and/or function of Treg cells in patients with SLE and murine SLE models.83–85
Several mechanisms have been proposed to explain defective IL-2 production in SLE. Reduced phosphorylation and expression of the TCR/CD3 ζ chain86 is one such mechanism. Two patients with SLE have been reported to have a 36-bp exon 7 deletion in the TCR ζ messenger RNA (mRNA), and many other mutations found in patients with SLE have been mapped to the third immunoreceptor tyrosine–based activation motif (ITAM) or the guanosine triphosphate/guanosine diphosphate (GTP/GDP)–binding site in the TCR ζ molecule. These mutations have been implicated in the downregulation of the TCR ζ chain.87 Transfection of SLE T cells with TCR ζ chain has been shown to normalize TCR/CD3-induced free intracytoplasmic calcium.86,88
Under physiologic conditions, the signal generated by the CD3 complex triggers phosphorylation of phospholipase C (PLC-γ) on Tyr and Ser residues, hydrolysis of phosphatidylinositol 4,5-biphosphate to phosphatidylinositol 1,4,5-biphosphate, and a rapid rise in intracellular Ca2+. The rise in intracellular calcium upon activation has been reported to be higher in SLE T cells than in control T cells.89 This increase in calcium flux in T cells, however, did not correlate with disease activity. The aberrant calcium flux is probably due to an IgG anti–TCR/CD3 complex antibody in human SLE serum.90 Tsokos has shown that this anti–TCR/CD3 complex antibody stimulates translocation of Ca2+ calmodulin kinase from the cytosol to the nucleus.86 This event induces upregulation of CREM (cyclic adenosine monophosphate [cAMP] response element [CRE] modulator) transcript and protein, phosphorylation of CREM and binding of pCREM homodimers to the −180 site of the IL-2 promoter, thus leading to decreased IL-2 production. Further studies suggest that protein phosphatase 2A (PP2A), the primary enzyme that dephosphorylates CREB (CRE binding) in T lymphocytes, is involved in the suppression of IL-2 production. Thus, PP2A represents a negative regulator of IL-2 promoter activity. Consistent with this idea, the mRNA, protein, and catalytic activity of PP2A are increased in patients with SLE regardless of disease activity and treatment.90
In contrast to the preceding studies showing increased calcium flux in SLE T cells, a study by Sierakowski found lower calcium flux upon anti-CD3 stimulation in patients with SLE than in controls.91 Ionomycin-induced calcium flux, however, is similar in patients with SLE and controls. The reduced calcium flux upon TCR stimulation in T cells was also seen in patients with mild disease and in those whose T cells produced normal amounts of IL-2.91 We have observed reduced calcium flux upon TCR signaling in T cells from autoimmune MRL/lpr mice at an age (≥8 weeks) when they begin to develop disease. Interestingly, T cells from these mice display a split activation phenotype—that is, although these T cells show evidence of in vivo activation as exhibited by increased expression of activation markers and IFN-γ production, they have reduced IL-2 production and calcium flux upon TCR stimulation (S Dubey and RR Singh, unpublished data, 2005).
Two cytoplasmic intracellular signaling pathways important in T-cell activation, differentiation, and effector function are the mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K). There are three major groups of MAP kinases in mammalian cells92: extracellular signal regulated kinases (ERKs), p38 MAP kinases, and c-Jun N-terminal kinases (JNKs). Defects in the MAPK signaling pathway in T cells have been shown to account for reduced IL-2 production by SLE T cells. For example, the activity of ERK-1 and ERK-2 is diminished in resting as well as TCR-stimulated peripheral blood T cells from patients with SLE; such a diminution can lead to reduced translocation of nuclear factor AP-1 (activator protein 1), resulting in altered coordination of signals needed for normal IL-2 production and maintenance of tolerance in T cells.93 Studies using the graft-versus-host disease (GVHD) model of murine lupus have found increased activity for PI3K and JNK but not for raf-1, p38 MAPK, or ERK-1. Increased PI-3 kinase activity in the chronic GVHD model is consistent with a role for persistent T-cell activation in lupus-like disease, as evidenced by increased phosphorylation of TCR-associated Src-family kinases (Lck and Fyn).94 Consistent with these data, treatment with a PI3 kinase inhibitor improves disease in MRL/lpr lupus mice.95 The PI3K pathway is also activated in peripheral blood mononuclear cells (PBMCs) and T cells from about 70% of patients with SLE, especially in patients with active disease. The magnitude of PI3K pathway activation in patients with SLE correlated with accumulation of activated/memory T cells. The study suggests that increased PI3K activity causes defective activation-induced cell death in patients with SLE. Moreover, defective activation-induced cell death in SLE T cells was found to be corrected after reduction of PI3Kδ activity, suggesting that PI3Kδ contributes to induction of enhanced memory T-cell survival in SLE.96
The mammalian target of rapamycin (mTOR), a key regulator of metabolic activity, and its major upstream activator, PI3K/AKT pathway, have been implicated in SLE pathogenesis.97 mTOR affects many immune cells, including monocytes and dendritic cells, and influences the cytokine milieu during an immune response. The PI3K/AKT/mTOR pathway is upregulated in lupus B cells and T cells. Importantly, the mTOR inhibitor, rapamycin, has been found to ameliorate disease in lupus mice and reduce disease activity in patients with SLE who had been treated unsuccessfully with other immunosuppressive medications.98,99 In T cells, rapamycin treatment alters the signaling through the TCR, which attenuates the inappropriate activation of autoreactive T cells in SLE. It has been shown that the TCR complex in SLE is different from normal T cells.100 In SLE T cells, CD3ζ is replaced by the FcR γ chain, and instead of recruiting Lck, these cells recruit Syk. Rapamycin-treated T cells are reported to exhibit increased levels of CD3ζ and Lck, thereby normalizing calcium flux and IL-2 production.101 Moreover, Syk is a downstream target of mTOR, and a Syk inhibitor, R788, ameliorates lupus nephritis.102 Additionally, stimulating T cells through the TCR in the presence of rapamycin in vitro promotes the generation of Treg cells.103 Thus, the spontaneous PI3K/AKT/mTOR signaling activity in pathogenic T cells might contribute to reduced Treg cell number and/or suppressive activity in patients with SLE and mouse models.84,85,104
T-cell abnormalities in lupus can also be explained by the alterations in lipid raft composition and dynamics. The organization of signaling molecules into discrete membrane-associated microdomains, called lipid rafts, is vital for regulation of T-lymphocyte activation pathways.105,106 Lipid rafts play a central role in signal transduction, in the immune response, and in many pathologic conditions on the basis of two important raft properties, their capacity to incorporate or exclude proteins selectively and their ability to coalesce into small domains. As has been reported, SLE T cells contain larger pools of lipid rafts than normal T cells and produce lipid rafts more robustly upon anti-CD3 treatment than normal T cells. These changes are accompanied by a qualitative alteration in the composition of lipid rafts in SLE. Whereas CD3ζ and LAT (linker of activated T cells) are uniformly distributed on the surface of normal T-cell membranes, these molecules are organized in discrete clusters on membranes of SLE T cells. Unlike lipid rafts from normal T cells, those from SLE T cells contain FcRγ and activate Syk kinase.107
The localization of Lck to lipid rafts is essential for normal TCR-mediated signaling. The Lck is significantly reduced in both lipid rafts and nonraft portions of T lymphocytes from patients with SLE. Reduced expression of Lck in lupus T cells occurs because of increased ubiquitination and subsequent degradation of Lck, so that T cells become unresponsive to TCR-mediated signals.108 These findings imply chronic in vivo activation of T cells in SLE. However, the direct pathogenetic implications of the reductions in Lck in lupus T cells as well as factors that regulate Lck homeostasis in lipid raft domains and cause degradation of Lck in lupus T cells remain to be clarified. Further studies have shown greater expression of raft-associated ganglioside GM1 in SLE T cells. CD45, a tyrosine phosphatase that regulates Lck activity, is also differentially expressed and its localization into lipid rafts is increased in SLE T cells. Such altered association of CD45 with lipid raft domains may regulate Lck expression in SLE T cells. The altered lipid raft occupancy is not induced by serum factors from patients with SLE, but cell-to-cell contact is required to activate proximal signaling pathways.109
Although most studies have focused on identifying genes associated with altered T-cell functions in SLE, epigenetic regulation of gene expression, such as histone acetylation and methylation, may also contribute to impaired SLE T cell function.110 In fact, treatment with histone deacetylase inhibitors, such as trichostatin A, which corrects these impairments and suppresses lupus in mice,111 holds promise for humans.






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


