Chapter 39 Hematology
Overview
Hematopoiesis: Regulation of Cellular Elements of Blood
Blood cell production normally takes place in the bone marrow, and all the circulating cellular elements of the blood arise from the level of a pluripotent hematopoietic stem cell (Fig. 39-1). Hematopoietic stem cells have two important properties, extensive self-renewal and ability to differentiate and mature into red blood cells, granulocytes (neutrophils, eosinophils, basophils), monocytes, platelets, and lymphocytes (T cells, B cells, and NK cells). Little is known about the molecular mechanisms that control self-renewal, but this ability of stem cells is critical to the success of treatments such as bone marrow transplantation. More is known about the factors that control hematopoietic differentiation and maturation.
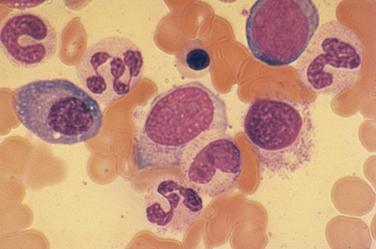
The regulation of the terminal stages of hematopoietic differentiation and maturation is under the control of a variety of hormones and cytokines. Red cell production is under the control of erythropoietin (EPO), a glycoprotein hormone secreted by the kidney in response to hypoxia or anemia. Neutrophil production is under the control of granulocyte colony-stimulating factor (G-CSF). Eosinophil production is under the control of interleukin-5 (IL-5). Monocyte and macrophage production is under the control of macrophage colony-stimulating factor (M-CSF) and, to a lesser extent, granulocyte-macrophage colony-stimulating factor (GM-CSF). Platelet production is under the control of thrombopoietin (TPO), produced in the liver. TPO also stimulates proliferation and release of hematopoietic stem cells. Of these various growth factors, EPO and G-CSF have important uses clinically; TPO is under clinical development. The number of cellular elements in the blood is tightly controlled by these regulatory hormones. Table 39-1 shows the normal ranges for the various cellular elements of the blood in adults.
Table 39-1 Cellular Elements of the Blood (Adult)
| Red Blood Cells (RBCs) | Men | Women |
|---|---|---|
| Hematocrit (%) | 46.0 ±4.0 | 40.0 ±4.0 |
| Hemoglobin (g/dL) | 15.7 ±1.7 | 13.8 ±1.5 |
| RBC count (×106/μL) | 5.3 ±0.7 | 4.6 ±0.5 |
| Reticulocytes (%) | 1.6 ±0.5 | 1.4 ±0.5 |
| Mean corpuscular volume (fl) | 88.0 ±8.0 | |
| Mean cell hemoglobin (pg/RBC) | 30.4 ±2.8 | |
| Mean cell hemoglobin concentration (g/dL of RBC) | 34.4 ±1.1 | |
| Red cell volume distribution width (RDW) (%) | 13.1 ±1.4 | |
| Platelets (/μL) | 150,000-300,000 | |
| White Blood Cells | Range (/μL) | Range (%) |
| All white blood cells | 4300-10,000 | 100 |
| Total neutrophils | 2000-7000 | 20-70 |
| Segmented neutrophils | 1500-6000 | 15-60 |
| Band neutrophils | 500-1000 | 5-10 |
| Lymphocytes | 1500-4000 | 15-40 |
| Monocytes | 100-900 | 1-9 |
| Eosinophils | 100-700 | 1-7 |
| Basophils | 0-150 | 0-1.5 |
Disorders of Red Blood Cells
The major role of the red blood cell (RBC, erythrocyte) is to transport oxygen from the lungs to the tissues and organs in the body. This is accomplished by the reversible binding of oxygen to the heme moiety of the hemoglobin molecule. Hemoglobin (Hb) is the most prominent RBC protein. The functional ability of the Hb molecule is determined by primary amino acid structure, pH (Bohr effect), temperature, and intracellular concentration of 2,3-biphosphoglycerate (2,3-BPG). Abnormalities of Hb function most frequently result from mutations (amino acid substitutions) in one or more of the globin genes. These mutations, such as the mutation that causes sickle cell disease, can alter the stability of the Hb molecule or its oxygen-binding properties. Many of these mutations result in tissue hypoxia because the instability of the molecule results in the early death of the RBC in circulation (hemolysis). Other Hb mutations can affect oxygen binding and release or can produce free-globin chains that may bind to the inner RBC membrane, resulting in membrane instability (Heinz bodies). Hemoglobin is also recognized as the major “sink” of nitric oxide (NO) in the blood. This has important implications for the control of vascular tone in the microcirculation.
Anemia
Definition
Anemia is best defined as a reduced RBC mass resulting in decreased oxygen-carrying capacity of the blood. Table 39-1 lists normal values for hemoglobin and hematocrit (Hct); values below the lower limits of normal for age and gender indicate only 25% likelihood that the person is not anemic. Previous Hb and Hct values for a given patient are useful comparisons when interpreting currently determined levels. Both pregnant women and long-distance athletes may have increases in plasma volume so that the Hct or the Hb value, or both, fall artificially below normal ranges. They still have a normal oxygen-carrying capacity with a normal RBC mass and should not be considered anemic. Conversely, a dehydrated patient admitted to the hospital may have Hb or Hct that appears normal because of plasma volume contraction. Correction of their volume-depleted state will allow appropriate determination of their true RBC oxygen-carrying capacity. When Hct or Hb is abnormal, further investigation is indicated, whether or not the subject appears clinically well.
Prevalence
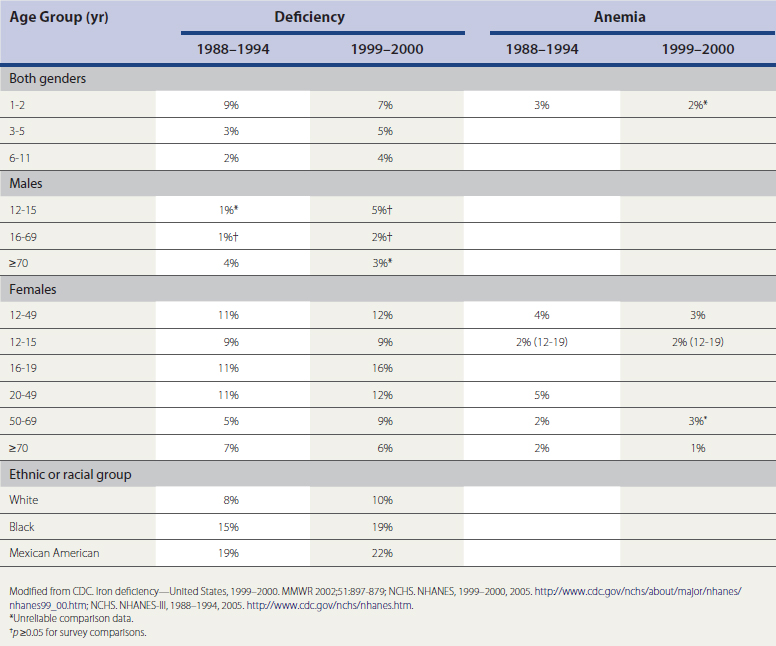
The most prevalent cause of anemia in the United States is iron deficiency. Table 39-2 categorizes the prevalence of iron deficiency and iron deficiency anemia (NHANES-III, 1988–1994, 1999–2000; NCHS, 2005a, 2005b). The prevalence of iron deficiency is 9% and of iron deficiency anemia 3%, in both genders age 1 to 2 years. The prevalence for children age 3 to 5 years is 3% for iron deficiency and less than 1% for iron deficiency anemia; those 6 to 11 years old have a 2% prevalence of iron deficiency and less than 1% for iron deficiency anemia. The prevalence of iron deficiency in the nonblack population is 1% lower than the prevalence in all races combined. Women have a much higher prevalence; those age 12 to 15 have 9% prevalence of iron deficiency and 2% for iron deficiency anemia; those age 16 to 19 have 11% and 3% prevalence; women 20 to 49 years, 11% and 5%; and those 50 to 69 years, 5% and 2%, respectively. In women over age 70, prevalence of iron deficiency rises to 7% and iron deficiency anemia to 2%. Males in general have less than a 1% prevalence of iron deficiency or iron deficiency anemia until age 50, when the prevalence of iron deficiency rises to 2%. In those older than 70, prevalence of iron deficiency is 4% and iron deficiency anemia 2%.
Clinical Features
Anemia is most often recognized by abnormal screening laboratory test results. Much less frequently, patients will present to their family physician with previously unrecognized anemia, complaining of fatigue, loss of stamina, shortness of breath, and rapid heart rate (particularly with physical exercise). In younger patients, if the anemia comes on gradually, several compensatory mechanisms help maintain tissue oxygenation. These include peripheral vasodilation, increased cardiac output, a change in the oxygen-hemoglobin dissociation curve that facilitates oxygen unloading in the tissues, and shunting of blood away from circulation-rich organs (e.g., gut, skin, kidney) to critical organs (e.g., heart, brain).
Laboratory Evaluation
Key laboratory tests in the office evaluation of the anemic patient include complete blood count (CBC), reticulocyte count, and iron studies (Box 39-1). The laboratory evaluation of anemia is designed primarily to determine effective response of the bone marrow to the anemia stimulus and to detect any disturbance in iron metabolism; this information allows physiologic classification of the most common anemias. The evaluation of the marrow’s response to anemia is best approximated through corrected reticulocyte count (Fig. 39-2), which provides information about the number of newly released RBCs in circulation. Normally, a newly released RBC can be seen as a reticulocyte for about 24 hours. The reticulum of a reticulocyte is made up of residual ribosomal ribonucleic acid (RNA); the cell is somewhat larger and appears bluer than mature RBCs on a Wright-Giemsa–stained peripheral blood smear.
Box 39-1 Laboratory Tests in Office Evaluation of Anemia
MCV, Mean corpuscular volume; MCH, mean cell hemoglobin; MCHC, mean cell hemoglobin concentration.
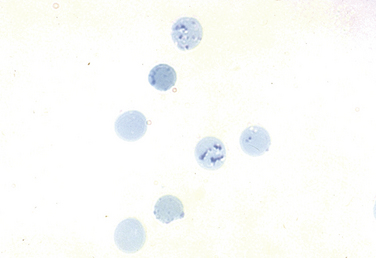
Figure 39-2 An increased number of reticulocytes are seen on a peripheral blood smear stained for reticulocytes.
(From the American Society of Hematology Image Bank image #1333. Copyright 1996 American Society of Hematology, used with permission.)
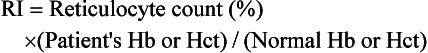
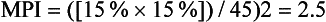
Also critical to understanding the pathophysiology of most anemias is to characterize the availability of iron for hemoglobin synthesis. This is done by measuring the serum iron level, total iron-binding capacity (TIBC), and serum ferritin level. Transferrin saturation percent is the proportion of iron binding sites occupied by iron atoms, reflecting the amount of iron immediately available for Hb synthesis, and is very labile. Serum ferritin is an indirect reflection of the body’s total iron stores and is more stable. These values provide information about the two most common forms of anemia seen in the hospital or in the family physician’s office—iron deficiency anemia and the anemia of chronic inflammation.
Physiologic Classification
The physiologic classification of anemia is based on response of the bone marrow. The three major categories are hypoproliferative anemia, maturation disorders (ineffective erythropoiesis), and hemolytic-hemorrhagic anemia (Box 39-2).
Hypoproliferative Anemia
In hypoproliferative anemias the response of the bone marrow is impaired by one of three general mechanisms. The first is marrow damage, which results from an injury to the bone marrow and makes it impossible for the marrow to respond to adequate EPO stimulation. Aplastic anemia is a classic example, but chemotherapy-induced marrow aplasia is more common (Box 39-3). The physiologic hallmark of marrow damage is a low RPI (<2 at Hb >10 g/dL; <2.5 at Hb of 7-10 g/dL). EPO is typically elevated, with evidence of premature release of reticulocytes from the bone marrow (shift reticulocytes, including nucleated RBCs in some cases). Despite EPO stimulation, however, the bone marrow is unable to proliferate. Bone marrow examination may reveal an empty or hypocellular marrow or one replaced by tumor cells or fibrosis. Treating these patients with recombinant human EPO is rarely useful.
The third major category of hypoproliferative anemia is that associated with inadequate EPO production, which results in understimulation of the marrow. Typically, this is seen in patients with chronic renal insufficiency whose diseased kidney cannot produce EPO despite often profound anemia. The advent of successful renal replacement therapy (peritoneal or hemodialysis) has resulted in an increasingly large population of severely and chronically anemic patients. For these patients, epoetin therapy has been lifesaving in terms of quality of life and overall health.
Mild iron deficiency anemia also is associated with a hypoproliferative marrow response. Iron deficiency, with mild to moderate anemia, impairs the erythroid marrow response. If the anemia is mild, circulating RBCs are normocytic or slightly microcytic, and the red cell distribution width (RDW) index is normal (Fig. 39-3). The serum iron is usually low, transferrin saturation less than 15%, and serum ferritin less than 15 ng/mL.
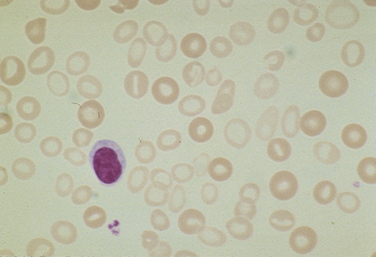
Figure 39-3 Hypochromic, microcytic red blood cells, with anisocytosis.
(From the American Society of Hematology Image Bank image #1214. Copyright 1996 American Society of Hematology, used with permission.)
Table 39-3 compares anemia of inflammation and classic iron deficiency anemia. The major difference is the serum ferritin level, which is typically normal or increased with AI and characteristically low with true iron deficiency. Making this distinction is important because the mechanisms that lead to inflammation or iron deficiency are generally distinct, as is the approach to treatment.
Table 39-3 Comparison of Anemia of Inflammation and Iron Deficiency Anemia
| Iron Deficiency | Anemia of Inflammation |
|---|---|
| Low serum iron level | Low serum iron level |
| Elevated TIBC | TIBC normal or reduced |
| Transferrin saturation low (<15%) | Transferrin saturation low (15%-20%) |
| Serum ferritin level low (<15 ng/mL) | Serum ferritin level normal or elevated |
| Microcytic, hypochromic RBCs | Normocytic to microcytic RBCs |
| RBC protoporphyrin level elevated | RBC protoporphyrin level elevated |
TIBC, Total iron-binding capacity; RBCs, red blood cells.
Maturation Disorders
Nuclear maturation defects result in a megaloblastic bone marrow and are typical in patients with severe folate or vitamin B12 deficiency. The RBCs are macrocytic, and the reticulocyte production index is normal or slightly above normal (Fig. 39-4). Examining the bone marrow of patients with vitamin B12 or folate deficiency reveals increased erythroid marrow precursors and loosening of nuclear chromatin, as well as more cells with nuclear degeneration (karyorrhexis and karyolysis). These are the features of apoptosis.
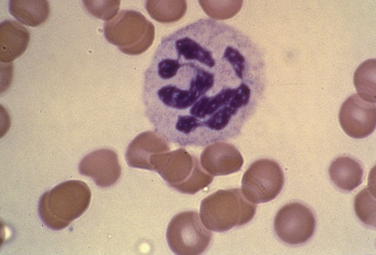
Figure 39-4 Megaloblastic changes of macrocytosis and hypersegmented neutrophils.
(From the American Society of Hematology Image Bank image #2611. Copyright 1996 American Society of Hematology, used with permission.)
Because of the degree of RBC destruction in the bone marrow, serum bilirubin may be elevated and haptoglobin decreased. It is important to distinguish between vitamin B12 and folate deficiency because the pathogenesis is different and the treatment must be specific. Patients who present with folate deficiency and alcoholic neuropathy pose a particularly difficult diagnostic challenge; the neuropathy of vitamin B12 deficiency should not be treated inappropriately with folic acid, because the anemia may be partly corrected with folic acid, but the neuropathy associated with vitamin B12 deficiency will progress. This is rarely seen at present. The neurologic symptoms may precede the anemia, so it is important to screen older adults with unexplained memory loss for vitamin B12 deficiency.
Thalassemia
Severe cytoplasmic maturation defects are usually inherited and are characteristic of the thalassemic syndromes or defects in heme synthesis. The inherited thalassemias represent a large number of mutations in the globin genes themselves or in the regulation of globin gene expression. When either the alpha or the beta globin chains are produced in unequal amounts, the excess chains aggregate and the erythroid precursor cells die, leading to ineffective erythropoiesis. β-Thalassemia produces decreased numbers of beta chains and α-thalassemia decreased alpha chains. Homozygous β-thalassemia is one of the most severe forms of human anemia; more than 200 million people worldwide carry the β-thalassemia gene. Because of the large number of mutations that can result in thalassemia, most patients who have the clinical phenotype of homozygous thalassemia are compound heterozygotes. Homozygous α-thalassemia is not seen in adults because it results in hydrops fetalis in the newborn. β-Thalassemia trait and α-thalassemia trait, however, are common but are usually associated with only mild anemia.
Hemolytic-Hemorrhagic Anemia
Intravascular Hemolysis
Mechanical fragmentation is probably the most common form of intravascular hemolysis seen in North America and is associated with microvascular diseases such as thrombotic thrombocytopenic purpura, hemolytic uremic syndrome (HUS; Fig. 39-5), defective mechanical heart valves, and disseminated intravascular coagulation. Acute attacks of malaria are also associated with intravascular hemolysis. Paroxysmal nocturnal hemoglobinuria (PNH) is an unusual form of intravascular hemolysis caused by an acquired X-linked defect in the hematopoietic stem cell. Patients with PNH have varying degrees of hemolysis throughout the day, and in crisis, this can be severe.
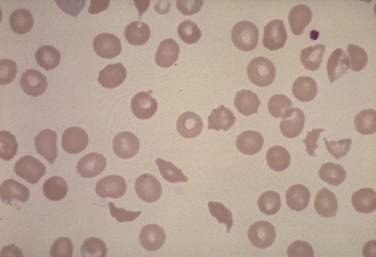
Figure 39-5 Schistocytes and helmet cells characteristic of a hemolytic process (hemolytic uremic syndrome).
(From the American Society of Hematology Image Bank image #4678. Copyright 1996 American Society of Hematology, used with permission.)
Extravascular Hemolysis
Contrasted with intravascular hemolysis is the destruction of RBCs that occurs primarily in the reticuloendothelial system (RES), so-called extravascular hemolysis. The primary site of destruction is outside the vascular compartment, in the liver, spleen, and bone marrow. A form of extravascular hemolysis is the ineffective RBC production seen with nuclear or severe cytoplasmic maturation defects. More common sites for extravascular destruction are the liver and particularly the spleen. Again, it is useful to determine whether the hemolysis is congenital or acquired. Congenital forms include inherited hemoglobinopathies (thalassemias, sickle cell disease), inherited membrane defects (hereditary spherocytosis or elliptocytosis), and enzyme defects. Defective RBC metabolism usually causes hemolysis by creating unstable Hb or by failing to generate adequate adenosine triphosphatase (ATP) to maintain RBC membrane plasticity, as occurs in pyruvate kinase deficiency; this autosomal recessive disorder is the most common enzyme deficiency of the glycolytic pathway. Most patients with pyruvate kinase deficiency have a mild anemia and generally do not require transfusions.
Sickle Cell Anemia
Clinical Diagnosis
Neurologically, patients with sickle cell anemia are subject to cerebrovascular accident (CVA, stroke). Particularly disturbing, children with severe sickle cell anemia will have evidence of multiple small strokes on magnetic resonance imaging (MRI) or computed tomography (CT) of the head by age 10 years. This has been associated with learning impairments in affected children. Aggressive transfusion protocols have been tested to reduce the incidence of this complication, with some success. Thrombosis is more common in children, whereas hemorrhage is more common in adults, possibly because of the occlusion of small vessels in hypoxic situations. Also, sickled cells show increased attraction to the endothelium, causing proliferation of the endothelial intimal surface, which can contribute to vascular occlusion. There is a high rate of recurrent stroke within 3 years of the initial event. Parvovirus infection can also cause pure RBC aplasia in these patients and lead to a devastating exacerbation of the anemia.
Polycythemia
In the evaluation of patients with polycythemia or suspected polycythemia, it is important to first establish that the RBC mass has increased. This is achieved through direct measurement of the red cell mass using isotope dilution techniques, and the test can be performed at most large hospitals. Once that issue has been resolved, and assuming that there is evidence of RBC overproduction, it is important to determine whether the polycythemia is primary or secondary by measuring the circulating EPO level. If elevated, causes of the increased EPO production to consider include heart or chronic pulmonary disease, in which there is desaturation of Hb as it leaves the lungs, or less common causes such as renal cysts, hepatic or cerebellar tumors, uterine leiomyoma, or impaired Hb function caused by heavy smoking, which results in elevated levels of carboxyhemoglobin, which is essentially inert as an oxygen transporter. Severe obstructive sleep apnea may cause enough desaturation to raise EPO levels, resulting in secondary polycythemia. Athletes using EPO, androgens, or blood doping may also present with secondary polycythemia.
Myeloproliferative Disorders
KEY TREATMENT






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


