Fig. 31.1
Acute cutaneous GvHD: characteristic morbilliform/maculopapular exanthema involving most of the body in a patient 30 days after allogeneic HSCT
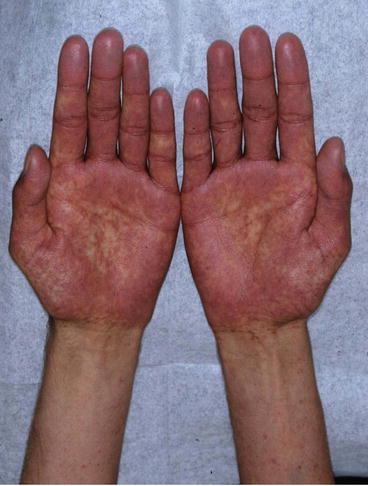
Fig. 31.2
Acute cutaneous GvHD: characteristic acral exanthema involving palms and soles in a patient 30 days after allogeneic HSCT
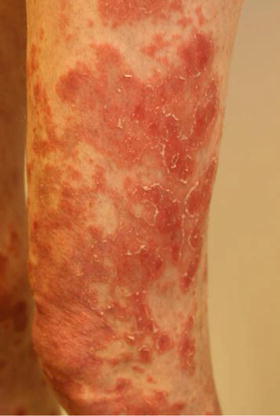
Fig. 31.3
Chronic cutaneous GvHD: disseminated erythemato-squamous plaques and patches involving most parts of the body in a patient more than 2 years after allogeneic HSCT. Clinicopathological correlation is consistent with overlap syndrome
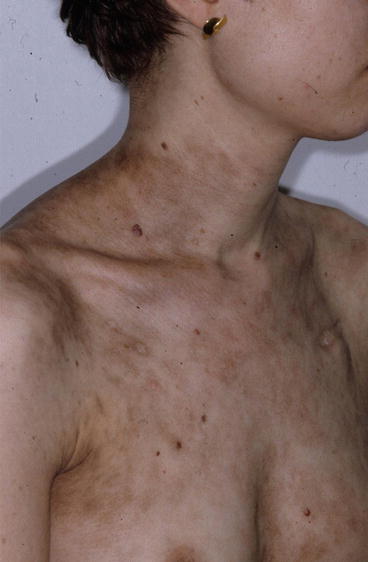
Fig. 31.4
Chronic lichenoid GvHD: characteristic livid and hyperpigmented patches in a patient 6 months after allogeneic HSCT
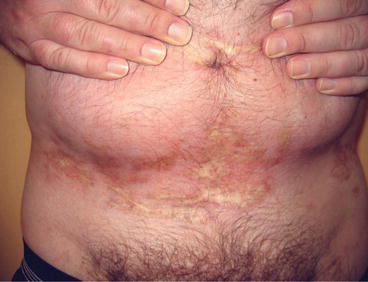
Fig. 31.5
Chronic sclerodermatous GvHD: characteristic sclerotic plaques in a patient more than 3 years after allogeneic HSCT
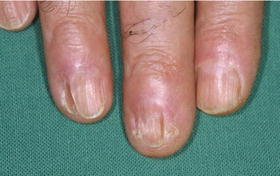
Fig. 31.6
Chronic cutaneous GvHD involving the nails: characteristic lichen planus-like changes of nails in a patient more than 2 years after allogeneic HSCT
In the past, acute and chronic GvHD have been separated stringently by time point after allogeneic HSCT. Any manifestations of GvHD present at ≥100 days posttransplant were arbitrary defined as chronic GvHD. As a result of newer conditioning and immunosuppressive regimens and multiple interventions (e.g., donor lymphocyte infusions), the distinction between simple categories such as acute and chronic GvHD is blurred. For instance, acute GvHD can be precipitated ≥100 days posttransplant by donor T-cell infusions to treat relapse and discontinuation of immunosuppressive medications (which initially prevented or treated acute GvHD). As a consequence, categories of acute and chronic GvHD were recently redefined by the NIH consensus development project groups. Acute GvHD is thus divided into classic (symptoms ≤ 100 days) and persistent, recurrent, or late-onset (symptoms ≥ 100 days) disease. Chronic GvHD is today categorized as classical (no time limit of symptoms, no presence of acute GvHD features) or overlap syndrome (no time limit, presence of acute and chronic features of GvHD) [6, 7].
Differential Diagnosis
In the appropriate setting of allogeneic HSCT, the differential diagnosis of acute cutaneous GvHD includes morbilliform drug exanthema, chemotherapy-related toxic exanthema, morbilliform viral exanthema (especially CMV), but also engraftment syndrome, eruption of lymphocyte recovery, or bacterial infections such as secondary syphilis. Depending on the predominant type of skin lesions and mucous membrane involvement, chronic cutaneous GvHD can imitate psoriasis, atopic dermatitis, drug-induced exanthema, vitiligo, or lichen planus. Often, the distinction to systemic autoimmune disorders such as lupus erythematosus, Sjögren’s syndrome, systemic sclerosis, or mixed connective tissue diseases can be challenging. Fibrosing disease is particularly reminiscent of lichen sclerosus, morphea, eosinophilic fasciitis (Shulman’s syndrome), and systemic sclerosis (limited or diffuse). Unlike systemic sclerosis, cGVHD does not manifest small vessel disease such as Raynaud’s phenomenon and nail typical fold capillaroscopy changes [5, 8].
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


