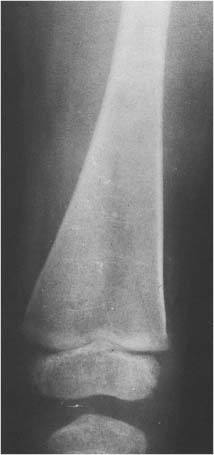
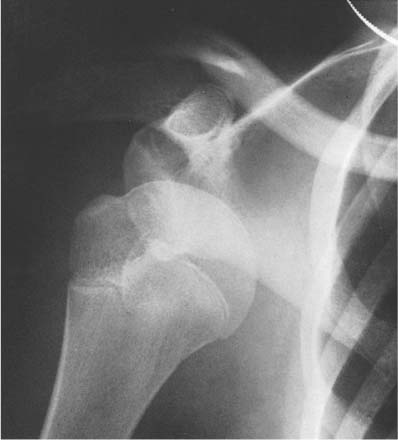
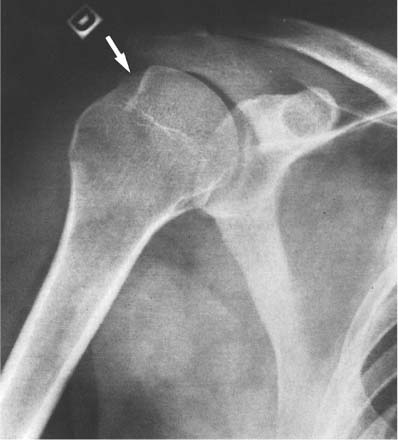
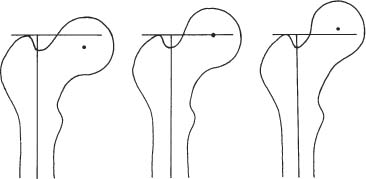
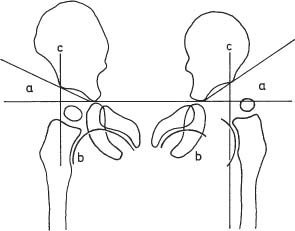
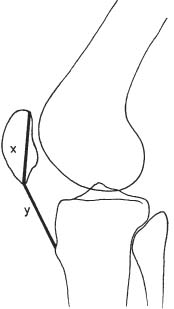
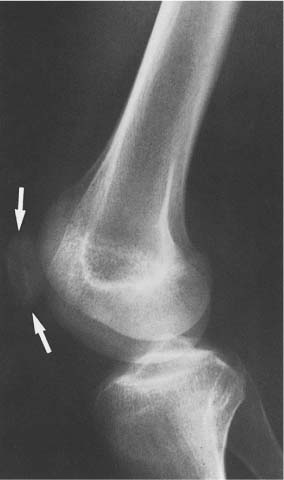
14 Extremities Deformities of the long bones are either developmental, that is, caused by abnormal or asymmetric growth, or they may be the sequelae of fractures or associated with a disease that causes bone softening. In the latter case, the weight-bearing lower extremities are usually more affected than the upper extremities. The differential diagnosis of long bone deformities is discussed in Table 14.1. Normally, the ends of long bones are wider than their diaphyses, and there is a progressive concentric decrease in the caliber of the shaft as one passes from the end toward the middle of a long bone. Abnormal modeling of the long bones can result from a variety of diseases affecting the growing bone and the end result is usually still evident in the adult. Less commonly, abnormal modeling of the long bones may only develop in adulthood. Such conditions are either associated with new periosteal bone formation (e.g. Paget’s disease or healing fractures and infections) or caused by expansile nondestructive mass lesions within the bone. A widening of the bone shaft is termed underconstriction or undertubulation. Widening or splaying of the metaphyses may be associated with underconstriction of the shaft or may present as an isolated finding. A splayed distal femur metaphysis often assumes the shape of an Erlenmeyer flask and is accordingly termed “Erlenmeyer flask“ deformity (Fig. 14.1). Conditions characterized by “Erlenmeyer flask” deformity include Gaucher’s disease, Niemann-Pick disease, thalassemia, craniometaphyseal dysplasia, Pyle’s disease, enchrondromatosis, hereditary multiple exstoses, osteogenesis imperfecta, osteopetrosis and lead poisoning. These disorders, as well as all other diseases with a more generalized, widened diametaphysis, are discussed in Table 14.2. Overconstriction (or overtubulation) is the reverse of underconstriction. As one passes from the end of a long bone toward the middle, there is excessive concentric narrowing of the shaft. Overconstriction is usually the cause of a chronic disease affecting the growing bone. It is only rarely acquired in adulthood by marked periosteal resorption of the cortex, since abnormal cortical resorption in the adult occurs predominantly on the endosteal margins, this does not lead to overconstriction. Table 14.3 deals with the differential diagnosis of overconstricted long bones. Transverse radiolucent and sclerotic bands or lines in the metaphyses are commonly found in the growing bones and do not necessarily indicate a pathologic process. They may persist into adult life. The differential diagnosis of radiolucent and sclerotic transverse bands is summarized in Tables 14.4 and 14.5, respectively. Long tubular bones continue to increase in length until fusion of the epiphyseal plates. Generalized elongation or over-growth of long bones is uncommon. The usual cause of over-growth in association with normal length-width relationships is gigantism, either of hypothalamic or pituitary origin. Elongation of bones with narrow diametaphysis (overconstriction, overtubulation) is seen in Marfan’s syndrome and homocystinuria. There are many individual variations in the width of bones, and it may be difficult to assess whether otherwise normal-looking bones are thin due to constitutional reasons or to a specific pathologic entity. If deformation of bone is an associated finding, as in neurofibromatosis, other clinical or radiographic signs are usually present to suggest a specific diagnosis. Generalized or localized shortening of a tubular bone is more likely to occur, since a number of congenital or acquired diseases or their treatment will interfere with the growth of bone. In Table 14.6, some conditions associated with generalized or localized shortening, hypoplasia, or deformity of the long bones of the extremities are presented. The majority of congenital bone dysplasias will not be mentioned here, since their differential diagnosis is more likely to be successful when based on the appearance of the hands and feet and the spine. The metaphyses of the long bones, especially the knee, hip, and shoulder regions, are favored sites of several malignant bone tumors. Also, benign bone tumors and tumor-like processes are common in the extremities. The general description of these diseases in Chapter 5 is based on their appearance in typical locations and will not be repeated here. Similarly, inflammatory lesions of bone such as bone infection, arthritis, and associated calcifications have been presented in a systematic fashion above. The femoral head is the most important site of fragmentation or deformation for traumatic, inflammatory, vascular, metabolic, or unknown reasons. Therefore, the causes of an abnormal proximal femur are presented in Tables 14.10–14.12, although some overlapping with other chapters unavoidably occurs. Many of the changes and diseases presented as typical of the femoral head also occur in other joints, especially in the shoulder. Fig. 14.1 “Erlenmeyer flask” deformity. The splayed distal tibia metaphysis assumes the shape of an Erlenmeyer flask (Pyle’s disease). Abnormalities of the alignment of the joints of the extremities may be the only sign of trauma, e.g., a dislocation without fracture. Malalignment may also be the result of a localized disease process characteristically seen in the knee joints. Since the alignment of the joint can be measured, it may be an objective starting point for a differential diagnostic approach, although it often represents a secondary phenomenon. Certain important facts concerning the alignment of the joints, and some terminological definitions, must be known in order to evaluate radiographs of the extremities appropriately and use Tables 14.7–14.13 properly. These will be presented here briefly. It is difficult to judge abnormalities of alignment in the shoulder (Table 14.7) because of its very wide range of normal motion. Shoulder dislocations are best detected from the axillary view (see Fig. 4.33). In anterior dislocation, the humeral head is displaced medially and inferiorly (superimposed over scapula, Fig. 14.2). A defect called Hill-Sachs deformity (Fig. 14.3), a compression fracture of the humeral head, is best seen when the arm is abducted and internally rotated 60 degrees. Fracture of the anterior inferior rim of the glenoid, known as the Bankart lesion, is less commonly seen. Bankart lesion may affect cartilaginous labrum only. Then the diagnosis can be made by MRI or by CT arthrography. Posterior dislocation of the humeral head is less common than anterior dislocation. It is difficult to recognize on a routine view. An abnormal appearance of the humeral head because of rotation may be the only sign of abnormality. In the axillary view the humeral head is superimposed on the acromion and does not articulate with the glenoid fossa. A compression fracture of the humeral head may be present. A fracture of the lesser tuberosity is so commonly associated with a posterior dislocation that its discovery on a routine view of the shoulder necessitates an axillary projection to determine its actual alignment. If the patient is capable of eversion of the humerus, the posterior dislocation is excluded. Dislocation inferiorly, towards the axilla, is rare and may be associated with a fracture of the greater tuberosity. Normally, the forearm makes an angle of approximately 170 degrees with the humerus, deviating laterally. Medial deviation (cubitus varus) and lateral deviation (cubitus valgus) of the forearm may result from trauma, connective tissue diseases, infections, neurotrophic diseases, and Turner’s syndrome. The most common malalignment in the hip (Table 14.12) is an abnormal angle between the shaft of the femur and its neck. Normally this angle is between 120 and 130 degrees in an adult. In children before puberty, values between 130 and 140 degrees are encountered. An increase in an adult above 130 degrees is called valgus deformity (coxa valga); a decrease below 120 degrees is a varus deformity (coxa vara). Valgus deformities are often related to congenital defects, while varus deformities may result from bone softening, such as osteomalacia or Paget’s disease. A simple method of determining if varus or valgus deformity is present is shown in Fig. 14.4. Normally there is anterior angulation (anteversion) of the femoral neck between 10 and 15 degrees in the adult, as compared with the line tangential to the distal femoral condyles. This angle is as high as 35 degrees in an infant and decreases towards adulthood. Without inversion of the inferior extremities (approximately 20 degrees), the angle of the femoral neck may not be correctly measurable in anteroposterior films of the hip. Various lines used for the determination of the congenital dislocation of the hip in a neonate are presented in Figure 14.5. When the tibia is angled laterally, too close approximation of the distal thighs results in knock knees (genu valgum). The opposite condition, separation of the distal thighs, is called bowlegs (genu varum). These deviations cause excessive stress on the knee and lead to degenerative changes. Medial displacement of the distal femur is called genu laxum. One measurement used to determine the position of the patella in a lateral view at flexion between 20 to 70 degrees is presented in Fig. 14.6. The ratio of maximal length of the patella to the distance from the distal tip of the patella to the tuberosity of the tibia is normally between 0.8 and 1.2. Patella alta (high patella) may be associated with recurrent subluxation, chondromalacia of the patella, rupture of the patellar ligament, osteomyelitis of the femur , poliomyelitis, avascular necrosis of the tibial tuberosity (Osgood–Schlatter disease), or the inferior ossification center of the patella (Sinding–Larsen–Johansson disease), and cerebral palsy. Patella profunda or baja (low patella) may occur as an inborn condition, as in achondroplasia, secondary to rupture of the quadriceps muscle tendon, or surgical transposition of the tibial tuberosity. It may also be secondary to juvenile rheumatoid arthritis or to paresis of the quadriceps muscle in poliomyelitis. Incongruity of patellar motion, e. g. lateral dislocation and tilting are more common and important abnormalities. Their demonstration is best done with cross sectional images using CT or MRI at flexion less than 30 degrees, since plain films are insensitive. Fig. 14.2 Anterior dislocation of the humeral head with characteristic medial and inferior displacement. Fig. 14.3 Compression fracture of the humeral head after dislocation (Hill-Sachs defect, arrow). The patella may be absent or hypoplastic. This is usually due to hereditary onycho-osteodysplasia or nail patella syndrome (Fig. 14.7), which is characterized by iliac horns and several other abnormalities. Hypoplasia of the patella is occasionally reported in association with acrocephalosyndactyly, and aplasia of the patella in association with bird-headed dwarfism, neurofibromatosis and popliteal pterygium syndrome. Fig. 14.4 a–c Evaluation of the angle of the femoral neck, a Coxa vara, b normal, and c coxa valga. Fig. 14.5 Determination of congenital dislocation of the hip in a neonate. The left side of the diagram represents normal relationships and the right side congenital dislocation, (a) Acetabular angle, (b) Shenton’s line, and (c) Ombredanne line. Fig. 14.6 Determination of the position of patella. The measurements should be done at flexion between 20 to 70 degrees, y/x is normally between 0.8 and 1.2. Patella alta = y/x > 1.2., patella profunda = y/x < 0.8. Fig. 14.7 Hereditary onycho-osteodysplasia. Hypoplastic patella (arrows). Table 14.1 Bowed or Deformed Long Bones
Alignment of Joints of Extremities
Disease | Radiographic Findings | Comments |
Congenital bowing | Bowlegs: Lateral and usually some anterior bowing of the proximal two-thirds of the tibias and/or the distal segments of the femora. Spurring of the medial aspect of the proximal tibia shaft or the posterior aspect of the distal femur shaft may also be present. | Idiopathic bowlegs of infancy disappear spontaneously after 2 to 3 years. Bowed limbs at birth may also be found in different forms of dwarfisms (e.g., achondroplasia and camptomelic and thanatophoric dwarfism). |
Osteodysplasty (Melnick-Needles syndrome) | Lateral bowing of the tibia characteristic. Sclerosis of the base of the skull and mastoids, tall vertebrae, ribbon-like appearance of the ribs, and metaphyseal flaring of short and long bones may also be evident. | Found only in females with small face, large ears, protruding eyes, micrognathia, malaligned teeth and short upper arms. |
Infantile cortical hyperostosis (Caffey’s disease) | Occasional sequelae of the disease in adulthood include bowing of the limbs, mandibular asymmetry and osseous bridging between adjacent bones. | The disease becomes evident in infants of less than 5 months of age. Usually the clinical and radiographic features subside slowly within a few months to a few years. |
Osteogenesis imperfecta (Fig. 14.8) | Bowing deformities in slender overconstricted and osteoporotic long bones with tendency to fracture characteristic. | Inherited connective tissue disorder. |
Hypophosphatasia | Bowing deformities and increased bone fragility are characteristically present both in the most severe form (seen in infants) and in the mildest form (seen in adults). In infancy, the disease resembles rickets (cupping and fraying of metaphyses), whereas in the adult the long bones are shortened and often demonstrate cortical thickening and pseudofractures. | Hypophosphatasia is a genetically determined metaphyseal dysplasia with failure to form primary spongiosa. Low serum alkaline phosphatase levels are characteristic. |
Vitamin D resistent rickets (x-linked hypophosphatemia) (Fig. 14.9a) | Bowing deformities occur in the long bones of the lower extremities. Pseudofractures (Looser’s zones) may be associated and progress to complete fractures. | Extensive enteropathy and a general increase in bone density (especially in the axial skeleton) are characteristic features in adulthood. In the spine the disease may simulate ankylosing spondylitis without sacro-iliac joint involvement or DISH. |
Madelung’s deformity (dyschondrosteosis) | Radius is bowed and short and its distal articular surface demonstrates ulnar and volar angulation. The distal ulna is deformed and subluxed dorsally. The carpal bones are wedged between the deformed radius and ulna and assume a triangular configuration with the lunate at the apex. May be associated with short and bowed tibia, short fibula, and valgus deformity of the knee. | Rare hereditary mesomelic bone dysplasia with female preponderance. Process usually begins in adolescence with premature fusion of the medial aspect of the distal radial epiphysis resulting in asymmetrical growth. In the majority of cases, the patients are of short stature and the deformities are bilateral. |
Blount’s disease (tibia vara) | Medial tibia condyle is enlarged and deformed with an irregular, beak-like extension originating from the media! metaphysis and projecting medially and posteriorly. Medial aspect of proximal tibia epiphysis may also be deformed. | Osteochondrosis of the medial tibia metaphysis resulting in bowlegs. An infantile type (usually bilateral) is differentiated from an adolescent type (usually unilateral). |
Weismann-Netter syndrome (Fig. 14.9b) | Bilateral and symmetrical anterior and medial bowing of the tibia and fibula, producing a “sabre-shin” deformity. The fibula shaft is typically thickened (“tibialization” of the fibula). | Congenital nonprogressive condition that has also been reported in the elderly. |
Enchondromatosis (Ollier’s disease) | Deformity and shortening of tubular bones containing round, often expansile lesions or alternating columnar radiolucent and radiodense streaks. Enchondromas may contain punctate or stippled calcifications. Disease may be relatively localized, unilateral, or generalized. | Disease is usually diagnosed in the first decade of life. May be associated with multiple hemangiomas containing phleboliths (Maffucci’s syndrome). In hereditary multiple exostoses bowing deformities typically are found in the radius and ulna. |
Osteomyelitis (Fig. 14.10) | Severe osteomyelitis may result in a deformed or bowed sclerotic tubular bone, particularly in the leg. Anterior bowing and cortical thickening of the tibia produces a “sabre-shin” appearance, characteristically found as late sequelae of both congenital and acquired syphilis and yaws. Bowing deformities confined to the distal half of the tibia associated with localized cortical thickening and solid periosteal reaction can be found in tropical ulcer. | Echinococcus rarely involves bones. Besides the spine, pelvis, and sacrum, the long bones may be affected with multiloculated cystic lesions, resulting occasionally in bone deformities. |
Bowing fracture | Anteroposterior or lateral bending of radius, ulna or fibula. A fracture in the neighboring bone (e.g. radius or tibia) might be associated. | Usually in young children. |
Fracture (healed) | Common cause of bone deformity usually associated with locally increased sclerosis. | All ages may be affected. Bowlegs in infancy are occasionally secondary to trauma. |
Paralysis during growth | Bowed long tubular bones with narrowed, over-constricted shaft diameter due to decreased medullary cavity while the width of the cortex is relatively little affected. | In longstanding paralytic and pseudoparalytic conditions such as old poliomyelitis, muscular dystrophy, and juvenile rheumatoid arthritis. |
Osteomalacia (Fig.14.11) | Bowing deformities occur, besides in the pelvis, spine, and thorax, particularly in the lower extremities. The cortex is thinned as in osteoporosis, but bone deformities, pseudofractures and poor definition of the inner cortical margin and trabeculae help to differentiate osteomalacia from osteoporosis. | Bowing deformities are also a common feature in rickets of any etiology. |
Hyperparathyroidism | Bone deformities similar to osteomalacia occur in an advanced stage. | In renal osteodystrophy (secondary hyperparathyroidism) bone deformities may be secondary to both osteomalacia and hyperparathyroidism. |
Paget’s disease (Fig. 14.12) | Bone deformities are common in the lower extremities involved by combined (sclerotic and lytic) phase of the disease. This results characteristically in coxa vara (“shepherd’s crook” deformity of the upper femur) and anterior bowing of the tibia. In this stage, the involved long bones are widened, and both cortex and disorganized-looking trabeculae are thickened. | Common cause of bone deformities in the elderly patient. Hereditary hyperphosphatasia (“juvenile Paget’s disease”), which occurs in children, presents characteristically with bowing and thickening of all long bones in the upper and lower extremities. |
Fibrous dysplasia (Fig. 14.13) | Marked deformities of the long bones occur, particularly in the polyostotic form, where the bone involvement is generally more extensive. “Shepherd’s crook” deformity (upper femur), cortical thickening, widening of the bone, and accentuated or abnormal trabeculation may be present, similar to Paget’s disease. The presence of radiolucent or cyst-like lesions or “ground-glass” appearance of the involved bone are useful in differentiating fibrous dysplasia from Paget’s disease. | In contrast to Paget’s disease, fibrous dysplasia is usually diagnosed in children and young adults. Skin pigmentations (“café-au-lait” spots with irregular outline) and sexual prematurity in females may be present in the polyostotic form (Albright’s syndrome). |
Neurofibromatosis (Fig. 14.14) | Overgrowth of the long bones may result in bowed appearance. The fibula frequently is slender (overconstricted). Pseudarthrosis (especially of the tibia) is common with this presentation. Bowed long bones may occasionally also demonstrate cortical thickening and increased sclerosis. | Hereditary disease often associated with skin neurofibromas and smoothly marginated “café-au-lait” spots. Congenital pseudarthrosis of the radius and ulna may also be associated with bowing, but this condition is not related to neurofibromatosis. |
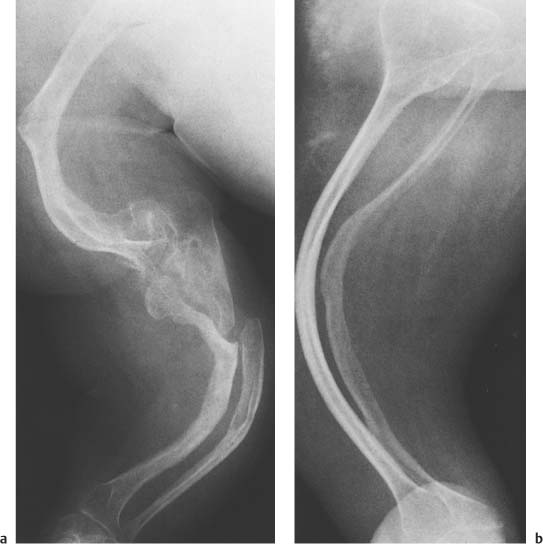
Fig. 14.8 a, b Osteogenesis imperfecta. a Upper extremity; b lower extremity. Bowing deformities in slender over-constricted and osteoporotic long bones are associated with fractures, pseudarthrosis (proximal ulna) and splayed metaphyses.
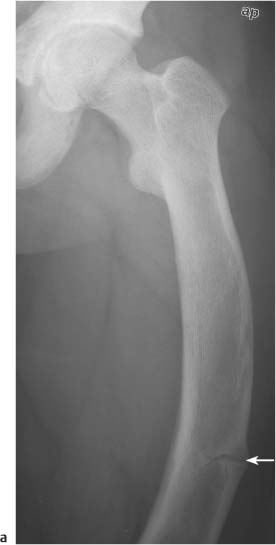
Fig. 14.9a Vitamin D resistent rickets (x-linked hypophosphatemia). Increased bone density is seen in this adult with lateral bowing of the femur and a pseudofracture (arrow) in the lateral cortex.
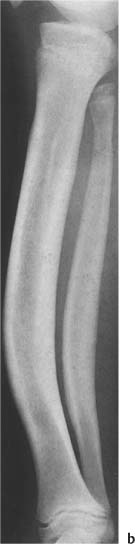
Fig. 14.9b Weismann–Netter syndrome. Anterior bowing of the tibia and fibula producing a “sabre-shin” deformity is seen. Note also the characteristic underconstriction of the fibula (“tibialization” of the fibula).
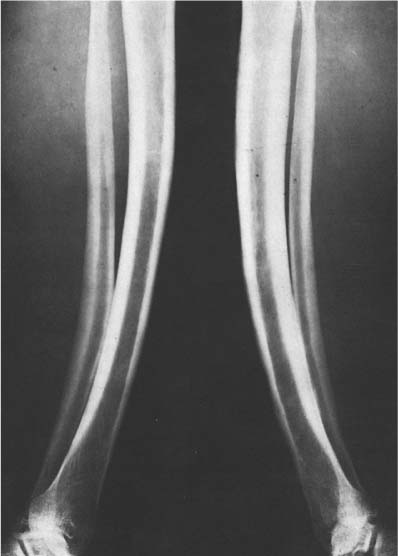
Fig. 14.10 Osteomyelitis (yaws). Cortical thickening and symmetrical anterior bowing of both legs produces “sabre-shin” appearance of both tibias as sequelae of the disease.
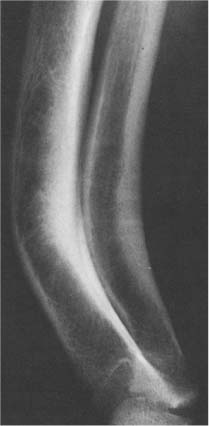
Fig. 14.11 Healed rickets. Late sequelae of severe rickets are seen in this adult with symmetrical bowing deformities of both legs including anterior and lateral bowing of the tibia and fibula and lateral bowing of the femur. Only lateral view of the right lower leg is shown.
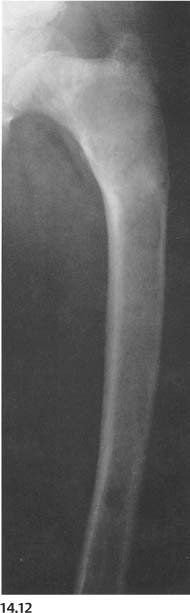
Fig. 14.12 Paget’s disease. Anterior bowing of tibia that is markedly widened, sclerotic, and irregularly trabeculated in its proximal two-thirds is seen.
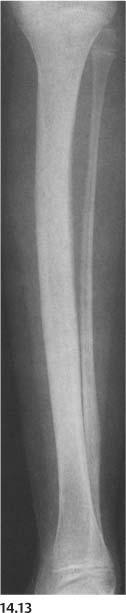
Fig. 14.13 Fibrous dysplasia. “Shepherd’s crook” deformity of the femur that is widened in its proximal half. Characteristic “ground-glass” appearance is also evident beside sclerotic changes and ovoid radiolucencies.
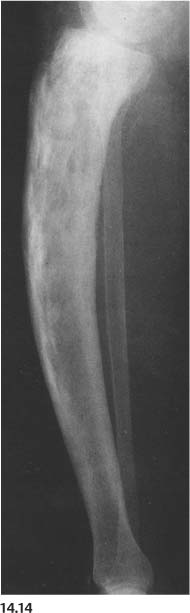
Fig. 14.14 Neurofibromatosis. Long slender (overconstricted) tibia and fibula with thickened cortex and slight medial bowing are present.
Table 14.2 Wide Diametaphyses (Underconstriction or Undertubulation)
Disease | Radiographic Findings | Comments |
Biliary atresia | Undertubulation and “Erlenmeyer flask” deformities combined with signs of osteoporosis and rickets are seen. | Congenital malformation resulting in progressive obstructive jaundice. Usually fatal within 2 years unless corrected surgically. |
Rubella embryopathy | Undertubulation of the long bones with alternating radiolucent and sclerotic longitudinal streaks (“celery-stick pattern”) and irregular metaphyses with radiolucent bands. | Osseous changes regress in those infants who grow normally, but may persist in those who fail to thrive. |
Infantile cortical hyperostosis (Caffey’s disease) (Fig. 14.15) | Periosteal new bone formation, cortical thickening and widening, primarily of the mandible, scapulae, clavicles, ribs and ulnae, are characteristic. | Disease develops in the first 5 months of life. Fever of abrupt onset, hyperirritability, and tender soft-tissue swelling over the affected bones are characteristic. The radiographic changes tend to regress spontaneously within one year. |
Progressive diaphyseal dysplasia (Engelmann-Camurati disease) (Fig. 14.16) | Symmetrical involvement of the long bones with undertubulation and cortical thickening of the midshaft resulting in a spindle-shaped appearing bone. | Rare hereditary disease associated with progressive midshaft cortical thickening and neuromuscular dystrophy. Usually diagnosed between 4 and 12 years, causing a wide-based, waddling gate. |
Mucopolysaccharidosis (Fig. 14.17) | Thickened undertubulated shafts, often with irregular wavy contours, are common findings. Metaphyseal flaring can be seen with Morquio’s disease, whereas tapering of the small tubular bones suggests Hurler’s disease. | Clinical and radiographic findings of different mucopolysaccharidoses and mucolipidoses are similar. A large dolichocephalic skull with J-shaped sella turcica, oval or hook-shaped vertebral bodies with gibbus formation in the thoracolumbar region, underdevelopment of the supra-acetabular region resulting in a widened acetabular roof, and coxa valga deformities with widened femoral necks, are characteristic. |
Metatropic dwarfism | Long tubular bones are shortened and have “dumbbell” appearance that is caused by the markedly enlarged metaphyses at both ends. | Short-limb dwarfism, which is normal at birth with progressive kyphoscoliosis, finally resembling Morquio’s disease. Flaring of the metaphyses with very short limbs are also seen in thanatophoric dwarfism, but this condition results in stillbirth or death in the neonatal period. |
Pseudoachondroplasia (Fig.14.18) | Initially the epiphyses are small and flattened and the metaphyses wide with dense, mushroom-shaped provisional zones ofcalcifications. In the adults the tubular bones are short and expanded at their end. | Spondyloepimetaphyseal dysplasias and epimetaphyseal dysplasias consist of a heterogeneous group of conditions that have metaphyseal flaring in common. |
Osteogenesis imperfecta (Fig. 14.19) | Metaphyses of the long bones are often markedly widened and may contain irregular calcifications while the shaft is overconstricted and may show severe bending deformities and multiple fractures of different ages. Osteopenia is usually a dominant feature. | Radiographic findings are more severe in the congenital than tarda form of osteogenesis imperfecta. |
Homocystinuria | Widened metaphyses and epiphyses with usually severe osteopenia and prominent growth lines are seen. In addition, Marfan-like changes (e.g., arachnodactyly) may be present. | Inborn error of the methionine metabolism. |
Pyle’s disease (familial metaphyseal dysplasia) (Fig, 14.20) | “Erlenmeyer flask” deformity or paddle-shapped enlargement of the metaphyses and, to a lesser degree, undertubulation of the long bones associated with cortical thinning and osteoporosis are characteristic. Thickened ribs and symmetrically enlarged proximal portions of the clavicles may be associated. | Rare hereditary disorder that can be differentiated from craniometaphyseal dysplasia by the lack of skull changes. |
Craniometaphyseal dysplasia (Fig. 14.21) | Undertubulation and “Erlenmeyer flask” deformities characteristic and usually most pronounced in the distal femur, radius, and ulna. Osteoporosis may be associated. Skull may show sclerosis of the base and calvarium, lack of aeration of paranasal sinuses and mastoids, and thickening and sclerosis of the mandible. | Rare congenital and familial bone dysplasia. Clinical features include hypertelorism, a broad flat nose, and defective dentition. |
Hereditary multiple exostosis (diaphyseal aclasis) (Fig. 14.22) | Undertubulation and often bowing of the long bones of both upper and lower extremities with multiple osteochrondromas in the metaphyseal regions, but usually sparing the elbow, are characteristic. Shortening of ulna and fibula is often present, Shortening of the ulna may result in congenital dislocation of the radial head, false articulation between the distal ulna and radius, and ulnar deviation of the carpus. | The inherited disease is usually diagnosed in the first decade of life. Transformation to chondrosarcoma occurs in approximately 5% of cases, although higher malignant transformation rates have been described. |
Enchondromatosis (Ollier’s disease) (Fig. 14.23) | Undertubulation, deformity, and shortening of the involved bones are common. Longitudinal radiolucent streaks interspersed with normal to sclerotic-appearing streaks may extend from the metaphysis into the shaft, or expansile radiolucent lesions with or without stippled calcifications may be seen. The metaphyses are usually most severly involved and often show “Erlenmeyer flask” deformities. | The generalized form of the disease is usually discovered in the first decade of life. The disease may also be unilateral or present only with a few enchondromas, characteristically located near a joint. Malignant degeneration in this nonhereditary disease is reported in 5–25% of cases. |
Bone cysts and benign tumors | Localized widening usually near the end of long bones is caused by a variety of benign expansile mass lesions. | For further differentiation, see Chapter 5. |
Chronic osteomyelitis (Fig. 14.24a) | Marked thickening of the involved area with scattered radiolucent, destructive lesions, solid periosteal new bone formation and sclerosis is characteristic. | In chronic sclerosing osteomyelitis (Carre), diffuse uniform sclerosis of the involved bone may be seen (Fig. 14.24b). |
Healed fracture | Localized to generalized widening of the shaft often associated with a bone deformity. Elevated solid periosteal reaction may initially simulate a double cortex or even “bone-within-bone” appearance during the healing phase, particularly in infants and children, but will disappear with further bone remodeling. Cortex may be thickened by periosteal bone formation, but may become normal or even thinned at a later stage. A metaphyseal injury may result in an “Erlenmeyer flask” deformity. | Common cause of localized undertubulation and deformity of a long bone. |
Thalassemia | Thinned cortices and a uniform reticular to cystic trabecular pattern in the widened diametaphyses with “Erlenmeyer flask” deformities characteristic, especially in the young patient. | A group of different hereditary hypochromic microcytic anemias, caused by the synthesis of abnormal hemoglobin polypeptide chains. Usually found in “Mediterranean” people and their descendants. Bone changes are caused by erythroid hyperplasia and are most pronounced in thalassemia major. Initially, all bones may be involved. With the conversion of red to yellow marrow in the tubular bones later in life the lesions may regress at these locations. |
Sickle-cell anemia | Cortical thinning and coarsening of the trabecular pattern due to bone marrow hyperplasia occur, but are less pronounced than in thalassemia. Changes in the long bones are caused by osteomyelitis and/or infarction and include destructive changes and new bone formation that may result in irregular diametaphyseal widening. Cortical infarcts may result in cortical thickening or “splitting of the cortex.” Layering of new bone along the inner cortex may produce a “bone-within-bone” appearance. | Sickle-cell anemia is a hereditary disorder limited almost exclusively to blacks, and caused by an abnormal hemoglobin HbS, where in position 6 of the β-globin chain, valine is substituted for glutamic acid. |
Gaucher’s disease (Fig. 14.25a, b) | Lower extremities are involved more often. Characteristic is the combination of avascular necrosis of the femoral head and an “Erlenmeyer flask” deformity at the distal femur. The shaft of the long bones may be locally expanded. Bone resorption and reparation may result in generalized osteopenia, lytic lesions of varying sizes simulating metastases, and patchy areas of sclerosis. Similarly, the cortex may be thinned, with a scalloped inner margin, or thickened and “split.” An inner layer of new bone formation may produce “bone-within-bone” appearance. Solid or lace-like periosteal reaction can occur, probably secondary to infarction. | Hereditary metabolic disease characterized by the accumulation of cerebrosides in the reticuloendothelial cells, resulting in hepatosplenomegaly. There is no sex predilection. The most common “adult” type occurs in Ashkenazic Jews, with the earliest manifestations appearing in childhood and progressing in the second and third decade of life. The acute type of Gaucher’s disease is a rare, fatal neurodegenerative disorder with no ethnic predilection, that usually becomes manifest shortly after birth and has an average survival time of one year. |
Niemann–Pick disease (Fig. 14.26) | Widening of the diametaphyses (undertubulation) and osteopenia with thinned cortices and coarsening of the trabecular pattern characteristic. Coxa valga occurs commonly. An “Erlenmeyer flask” deformity in the distal femur may simulate Gaucher’s disease. | Hereditary metabolic disorder characterized by the accumulation of phospholipids (sphingomyelins) in the reticuloendothelial cells. Niemann-Pick disease can be differentiated radiographically from Gaucher’s disease by the usual absence of avascular epiphyseal necroses, bone infarcts, and well-circumscribed radiolucent lesions. |
Langerhans cell histiocytosis (histiocytosis X) | Expansile lytic lesion and periosteal new bone formation may produce irregular localized widening of the long bones. |









