Chapter 5 Epigenetics of Lupus
For a long time, genetic variation has been thought to be the primary cause of systemic lupus erythematosus (SLE; also called lupus). This belief led to gene-hunting studies to identify a list of genes, such as the MHC region, IRF5, ITGAM, STAT4, BLK, BANK1, PDCD1, PTPN22, TNFSF4, TNFAIP3, SPP1, some of the Fcγ-receptors, and several complement components,1 which are well-established risk factors predisposing to lupus. However, genetic variation could not fully explain the pathogenesis of SLE. Environmental factors also influence the pathogenic processes.2 Now more evidence has emerged to support that epigenetic variation also plays a part in diseases in which environmental and genetic factors are both involved, for example, cancer and autoimmune diseases.3
Epigenetics refers to the inheritance of variation without changes in the DNA sequence.4 Up to now, studies on the epigenetics of SLE have focused on DNA methylation, histone modification, and microRNA (miRNA) regulation.
DNA Hypomethylation in SLE
DNA methylation usually occurs at the 5′ position of cytosine residues located in dinucleotide CpG sites that nonrandomly distribute in genomes.5 CpG-rich regions called CpG islands, about 500 to 5000 base pairs (bp) long, usually extend in the promoter and the first exon of genes. Other lone CpG dinucleotides are located in the intergenic and intronic regions, particularly within repeat sequences and transposable elements.5 DNA methylation patterns are regulated by particular methyltransferases, namely, DNA (cytosine-5)-methyltransferase 1 (DNMT1), DNMT3A, DNMT3B, and DNMT3L.6,7 DNMT1 maintains DNA methylation by replicating existing methylation patterns. DNMT3A and DNMT3B establish de novo DNA methylation. DNMT3L assists the function of DNMT3A and DNMT3B8,9 but does not contain any intrinsic DNA methyltransferase activity.8 In normal somatic cells of humans, 70% to 90% of CpG dinucleotides are methylated.10,11 Conversely, abnormalities in DNA methylation can lead to increased or decreased expression of genes and transposable elements, which may contribute to disease.5
It has been reported that the DNA methylation level is lower in thymus and axillary lymph nodes of diseased 20-week-old MRL/lpr mice.12 In humans, DNA extracted from the T cells of patients with lupus is hypomethylated compared with the DNA from normal T cells.13 Various environmental factors, such as procainamide, hydralazine, ultraviolet (UV) light, aging, and diet, can prevent the replication of DNA methylation patterns during mitosis, resulting in the DNA demethylation in T cells and lupus-like autoimmunity.14–16 Such agents usually induce the overexpression of autoimmune-associated methylation-sensitive genes, such as TNFSF7 (CD70) and LFA-1, which confer an autoreactive status to T cells.17,18 Adoptive transfer of T cells made autoreactive by treatment with DNA methylation inhibitors or by transfection with LFA-1 is sufficient to cause a lupus-like disease in unirradiated syngeneic mice.19,20 All of these findings suggest that DNA hypomethylation plays a crucial role in the pathogenesis of SLE. However, mechanisms that may contribute to low levels of T-cell DNA methylation in SLE remain to be studied. It has also been reported that miRNAs, such as miR148a21 and miR126,22 and the ERK signaling pathway15,23 can regulate DMNT1 levels in T cells from patients with SLE. Despite the regulation of DNMT1 expression, other observations suggest that DNA demethylation may also play a role.24 The p53-effector gene GADD45a, which may participate in DNA demethylation, has a higher expression level in CD4+ T cells of patients with SLE than in normal people. Moreover, UV light can induce GADD45a expression, and GADD45a−/− mice can demonstrate SLE-like autoimmunity disease.25
Histone Modification Changes in SLE
The nucleosome, basic unit of chromatin, is composed of a histone octamer (H2A and H2B dimers and H3/H4 tetramers) surrounded by 146 bp of DNA. Tails in the N end of histones protruding outside the octamer have different posttranslational modification, including acetylation, methylation, ubiquitination, phosphorylation, sumoylation, and adenosine diphosphate (ADP) ribosylation. These modifications can change the interaction between histone and DNA so as to affect DNA replication, transcription, DNA repair, and chromatin relaxation or condensation.26
In general, some histone modifications have certain association with gene expression activation or repression. For example, H3 and H4 hyperacetylation (H3Ac, H4Ac), H3 trimethyl-lysine4 (H3K4me3), H3 trimethyl-lysine36, and H3 trimethyl-lysine72, are present in many active genes; while H3 and H4 hypoacetylation, H3 trimethyl-lysine9 (H3K9me3), H3 trimethyl-lysine27 (H3K27me3), and H4 trimethyl-lysine 20 (H4K20me3), are characteristic of many repressed genes and heterochromatin.28 Like DNA methylation, the balance of histone modifications is also established and maintained by a group of enzymes, such as for histone lysine acetyltransferases and demethylases, histone lysine and arginine methyltransferases and demethylases, histone serine phosphorylases. Now many of these enzymes are becoming potential targets for the development of new therapeutic compounds.
However, the role of histone modifications in the pathogenesis of SLE is not well understood. Although examination of the global histone modification pattern in both MRL−lpr/lpr mice splenocytes and CD4+ T cells from patients with SLE showed H3 and H4 hypoacetylation and site-specific histone methylation changes,27,28 the roles of these modification variations in the process of SLE pathogenesis are not clear. One study reported that the histone deacetylase inhibitor trichostatin A (TSA) can restore skewed expression of CD154 (CD40L), interleukin (IL) 10, and interferon gamma (IFN-γ) in lupus T cells.29 Similarly, treating MRL−lpr/lpr mice with TSA and suberoylanilide hydroxamic acid (SAHA), another histone deacetylase inhibitor, decreased expression of IL-6, IL-12, IL-10, and IFN-γ and modulated renal disease through reduction in proteinuria, glomerulonephritis, and spleen weight.30,31 This finding suggests that histone modification variation can also play a role in lupus and offers a potential new way to treat this disease.
microRNAs in SLE
microRNAs (miRNAs) are a novel class of endogenous, noncoding small RNAs 19 to 25 nucleotides in length. They are ubiquitous in a wide range of species, such as viruses, worms, flies, plants, and animals,32,33 and function to negatively regulate gene expression at the posttranscriptional level. Although our current knowledge of miRNAs is still limited, it is being gradually accepted that miRNAs can modulate gene expression similarly as the transcription factors (TFs) in higher eukaryotes, representing a new layer of gene regulation. In parallel, mixed regulatory circuits are emerging in which close interplay between miRNAs and TFs cooperatively contributes to the formation of a complex posttranscriptional network.34 It has been well established that miRNAs are involved in multiple physiologic and pathologic processes, including stem cell development, cell differentiation and organogenesis, proliferation and apoptosis, immune regulation, and disease development.32,35–37
miRNA Biogenesis
Genomic analysis of miRNA transcripts38,39 revealed that a large proportion of miRNAs reside within introns of coding or noncoding regions, with a few in exons of long noncoding regions. Generally, miRNA genes are transcribed by RNA polymerase II to generate stem-loop primary miRNAs composed of one or several miRNA hairpin structures.40 The primary miRNAs (also called pri-miRNAs) are sequentially recognized by DiGeorge syndrome–critical region gene 8 (DGCR8), which functions, by formation of a microprocessor complex with nuclear RNase III enzyme Drosha, to produce pre-miRNAs (or miRNA precursors). After being actively transported to cytoplasm via the exportin-5 pathway, the pre-miRNAs are further processed by RNase III Dicer to yield the miRNA duplex, the “mature” miRNA. One functional strand of this duplex is then recognized by Argonaute (Ago)–containing RNA-induced silencing complex (RISC) and loaded onto the messenger RNA (mRNA) target with imperfect complementarity.40 This leads to either destabilization (most miRNA impact falls into this category41) or translational repression of target mRNAs.42–44 In some relatively rare cases, intronic miRNAs, called mirtrons, can bypass Drosha processing and be processed only by Dicer as pre-miRNAs.45–47 In addition, it has been reported that maturation of the microRNA miR-451 can be directly processed by Ago2, an indispensable catalytic component of RISC, without the participation of Dicer.48 Intriguingly, it has also been reported that anti-Su antibodies in sera from human patients with rheumatic diseases can recognize Ago2 and Dicer, two core catalytic enzymes in the miRNA pathway,49 but the effect of these autoantibodies on miRNA biogenesis is not clear.
The biogenesis of miRNAs is a highly regulated process that involves participation of multiple proteins at various stages. The maturation of miRNAs might be the key regulatory step in miRNA biogenesis, which can be well exemplified by the deliberately controlled maturation of the tumor suppressor miRNA let-7 (Figure 5-1). By impairing the Drosha-mediated pri-miRNA processing step, the nuclear factor NF90-NF45 complex negatively regulates Let-7 biogenesis.50 In contrast, the KH-type splicing regulatory protein (KSRP) promotes this processing by binding to the terminal loop of the pri-let-7.51 Heteronuclear ribonucleoprotein A1 (hnRNP A1), another negative regulator, blocks the pri-let-7a processing through antagonizing KSRP-binding activity.52 In addition, Lin-28/28B, as a highly conserved RNA-binding protein, exerts an inhibitory effect on let-7 maturation through inhibition of pri-let-7 processing and pre-let-7 cleavage mediated by Drosha and Dicer,53–55 respectively. One study has reported that miR-107 regulates let-7 stability through direct interaction with it, thereby participating in cancer progression and metastasis.56
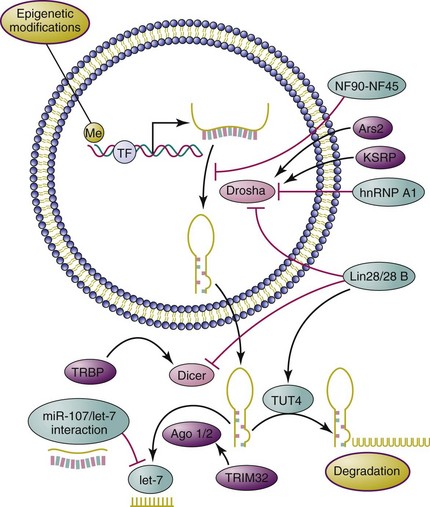
Novel Functions of miRNA in the Immune System
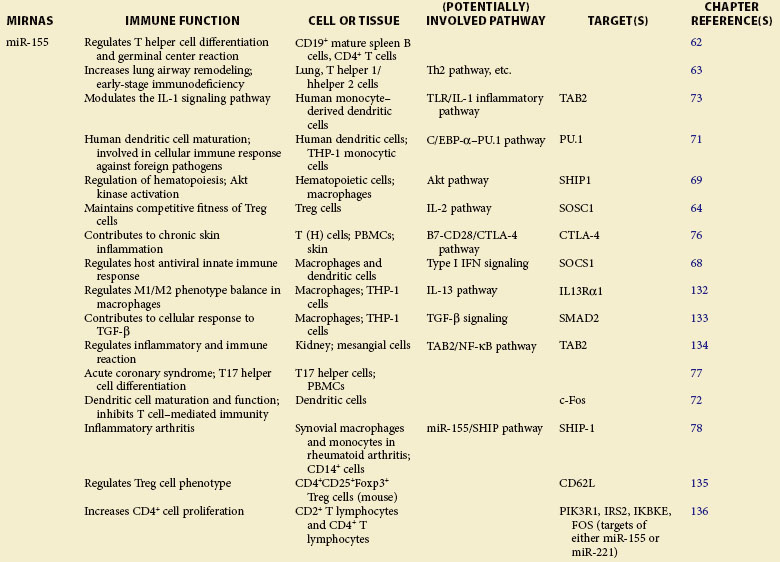
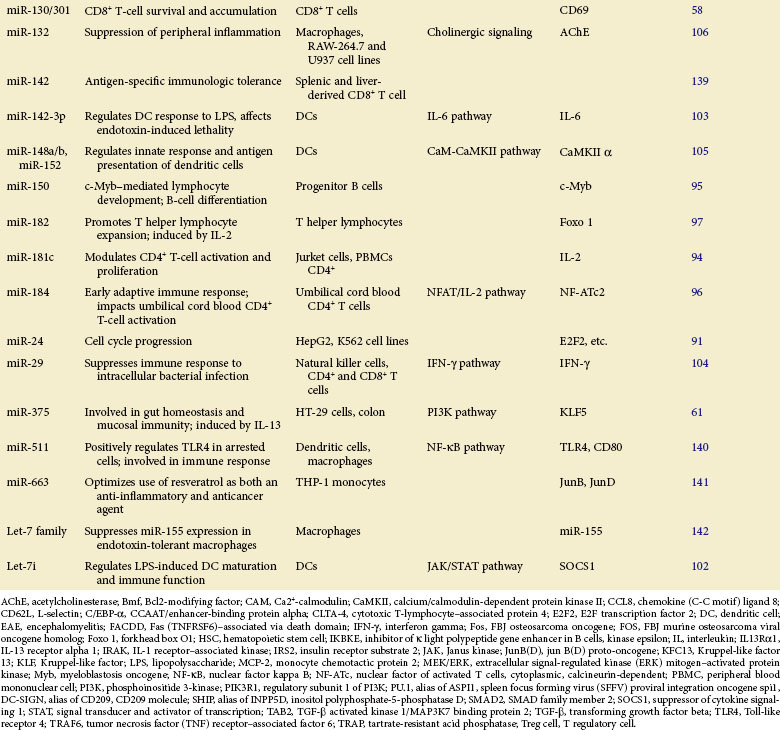
Table 5-1 summarizes the functions described here.

Dicer−/−
Genetic ablation of the key component involved in miRNA biogenesis can severely impair immune development and response. Depletion of Dicer protein, a crucial miRNA-processing RNaseIII enzyme, causes disrupted Regulatory T (Treg) cell–mediated tolerance,57 impaired CD8+ T-cell survival and accumulation,58 and blocked progenitor B-cell differentiation.59 In human leukemic cells deficient in Dicer, significantly enhanced apoptosis has also been observed.60 Specific Dicer1 deletion in gut epithelium renders mice more susceptible to parasites as a result of ineffective immune response.61 Mice deficient in Dicer in peripheral mature CD8+ T cells showed reduced T-cell expansion and immune response upon infection.58 These findings indicate a pivotal role for Dicer and its mediated RNA interference (RNAi) machinery in normal immune system maintenance.
miR-155
Depletion of miR-155 Causes Severe Immune Deficiency
A role of miR-155 in the immune system was first demonstrated in bic/miR-155−/− mice.62,63 Through specific regulation of T-cell differentiation and germinal center response, miR-155 influences the T cell–dependent antibody generation and controls lymphocyte cytokine production—tumor necrosis factor alpha (TNF-α), lymphtoxins alpha and beta (LT-α/β, interleukins 4 and 10 (IL-4/10), interferon gamma (IFN-γ ), and so on.62 Although lymphoid cells from miR-155–deficient mice exhibited normal cell development, lymphocyte immune deficiencies such as altered T helper 1 cell (Th1) function, skewed Th2 differentiation, and defective B-cell class switching, were observed.62,63 In addition to the effect of miR-155 on differentiation and immune function of T and B cells, recent studies extend its role in Treg cell regulation. It was reported that miR-155 regulates Treg cell homeostasis by specifically inhibiting expression of suppressor of cytokine signaling 1 (SOCS1), a key negative regulator of cytokine signaling.64 This miRNA was also shown to be critically involved in Treg cell–mediated tolerance through regulation of CD4+ Th cell activity, in which depletion of miR-155 resulted in enhanced cell sensitivity to natural Treg (nTreg)–mediated suppression.65
miR-155 Is a Multifunctional Regulator in Toll-Like Receptor Signaling
The robust upregulation of miR-155 upon stimulation of multiple Toll-like receptor (TLR) ligands66,67 indicates the role of miR-155 in response to bacterial and viral infection. Indeed, numerous molecular targets have been identified for miR-155 in TLR signaling. In IFN-mediated antiviral response, SOCS1 is targeted by miR-155 in macrophages to attenuate viral propagation.68 Inositol-5′-phosphatase SHIP1 (alias of INPP5D, inositol polyphosphate-5-phosphatase D), which negatively regulates TLR4 signaling, can be targeted for repression by miR-155 induced in response to lipopolysaccharide (LPS) stimulation.69 miR-155 is also critical for dendritic cell (DC) maturation and its antigen-presenting cell (APC) function.70 Transcription factors PU.171 and c-Fos72 have been identified as direct targets of miR-155, the repression of which leads to functional defects in DCs. In LPS-activated DCs73 and plasmacytoid DCs,74 TAK1-binding protein 2 (TAB2), an essential molecule that regulates TLR-mediated nuclear factor kappa B (NF-κB) activation by recruiting TRAF6, has been confirmed as a direct target of miR-155. In addition, miR-155 was demonstrated to repress the expression of MyD88,75 a vital adapter molecule in TLR signaling.
Involvement of miR-155 in Inflammation
miR-155 has been reported to be potentially implicated in the pathogenesis of multiple inflammatory disorders, including atopic dermatitis,76 acute coronary syndrome,77 and inflammatory arthritis.78 miR-155−/− mice exhibit strong resistance to experimental autoimmune encephalomyelitis induced by myelin oligodendrocyte glycoprotein 35-55 (MOG35-55), with defective inflammatory T-cell development, mass loss of Th17 cells, and markedly reduced production of Th17-relevant inflammatory cytokines.79 In a study of skin inflammation, upregulation of miRN-155 was observed in activated T cells, resulting in repression of cytotoxic T-lymphocyte–associated protein 4 (CTLA-4) in T cells.76 miR-155 is also required for the development of collagen-induced arthritis; stronger resistance to the disease was reported in miR-155 mutant mice that had reduced proinflammatory cytokine production.78
miR-146a: A Critical Immunomodulator
In 2006, Taganov first reported that miR-146a is highly induced upon LPS stimulation, as a strong negative regulator of TLR signaling, in human monocytes with targeted repression of TNF receptor–associated factor 6 (TRAF6) and interleukin-1 receptor–associated kinase 1 (IRAK1).80 It was later shown that miR-146a can be induced by various inflammatory ligands in monocytic THP-1 cells, the expression of which is inversely correlated with TNF-α production, rendering cells tolerant81 and cross-tolerant82 to TLR stimulus. These results were further confirmed by an in vivo study demonstrating that the sustainably expressed miR-146a induces proteolytic degradation of IRAK1 during the neonatal period, contributing to innate immune tolerance of the intestinal epithelium.83 miR-146a was also reported to be highly expressed in Treg cells and to selectively regulate Treg-mediated suppression, which inhibits IFN-γ–dependent Th1 activity and inflammation, by acting on transcription factor STAT-1 (signal transducer and activator of transcription 1).84 Moreover, an elevation of miRNA-146a was observed in HIV-infected microglia cells and brain specimens from patients with HIV encephalitis (HIVE), in which the chemokine CCL8/MCP-2 was identified as its specific target.85
Apart from its role in innate immune response, miR-146a has also been reported to function to modulate adaptive immunity and participate in disease pathogenesis. Curtale reported that the expression of miR-146a can be affected by T-cell receptor (TCR) signaling activation, and its induction leads to impairment of IL-2 production through modulation of AP-1 (activator protein 1) transcriptional activity.86 In addition, high miR-146a induction was reported to reduce the expression levels of transcription factors Jun, NF-ATc1, PU.1, and TRAP in peripheral blood mononuclear cells (PBMCs), resulting in alleviation of bone destruction in rheumatoid arthritis (RA).87
Other miRNAs
miRNAs in Immune Cell Differentiation and Maturation
In mammals, it has been well established that miRNAs function as positive modulators for hematopoietic lineage differentiation.88,89 One study has revealed that highly expressed miR-125b in hematopoietic stem cells (HSCs) promotes their differentiation toward lymphoid lineage.90 It was also demonstrated that miR-24 is upregulated and functions in hematopoietic cell terminal differentiation by targeting E2F291 and H2AFx,92 two key components regulating cell cycle progression.
It has been shown that miR-181, which is highly expressed in spleen and thymus, plays a crucial role in both B- and T-cell differentiation. Transplantation of lethally irradiated mice with bone marrow cells overexpressing miR-181a results in increased CD19+ B-cell proliferation along with severe CD8+ T-cell reduction.88 When ectopically expressed in undifferentiated B-cell progenitors, miR-181a specifically promotes B-lymphocyte differentiation in mouse bone marrow,88 whereas its upregulation in immature T cells is responsible for modulating TCR signaling (positive and negative selection) by inhibiting expression of multiple downstream phosphatases,93 thereby negatively regulating the downstream signaling cascades. Another study has revealed that ectopic expression of miR-181c inhibits IL-2 expression and reduces cell proliferation in activated CD4+ T cells.94
The roles of miRNAs in regulation of lymphocyte development are often achieved by posttranscriptional negative regulation of key transcription factors in corresponding signalings. For example, miR-150 is specifically expressed in mature B cells, but not in their progenitors. The ectopic expression of miR-150 in mice dramatically impairs B-cell development with a remarkable reduction in B1 cell numbers via targeted repression of c-Myb protein, a critical transcription factor required for pro–B-cell differentiation.95 In the early adaptive immune response, miR-184 influences immune cell activation (umbilical cord blood CD4 T cells) and limits downstream IL-2 production by targeting NFAT1, a key transcription factor regulating the production of multiple proinflammatory cytokines.96 Moreover, it was reported that during T helper lymphocyte development, miR-182 expression is induced by IL-2 to inhibit transcription factor Foxo 1 activity, resulting in T-cell clonal expansion.97
miRNAs in Immune Response
The roles of miRNA in regulation of immune response has been extensively investigated over the past several years.32,35–37 Numerous miRNAs have been identified with functions intimately related to TLR signaling.98 Later studies provide more evidence for the notion that miRNA plays an essential role in control of immune cell function through precise modulation of key molecules in TLR signaling for prevention and control of excessive inflammation.
For example, dust-induced TLR activation and the consequent inflammatory response in allergic asthma can be inhibited by miR-126, which indirectly reduces expression of the PU.1 and transcription activator OBF.1/BOB.1.99 Also, miR-124 was shown to be involved in the inhibition of macrophage activation and attenuation of central nervous system (CNS) inflammation by repressing expression of transcription factor C/EBP-α (CCAAT/enhancer-binding protein alpha) and limiting the production of TNF-α in macrophages.100 It is noteworthy that the same miRNAs, in different cellular contexts or under distinct pathologic conditions, may not act uniformly when immune cells develop immunity to foreign pathogens. In human cholangiocytes, for example, let-7i, which functions to negatively regulate TLR4 expression, is downregulated in response to LPS stimulation and bacterial infection.101 In LPS-induced DC maturation, however, let-7i was reported to be upregulated to maintain LPS-induced production of proinflammatory cytokines (IL-12, IL-27, TNF-α, and IFN-γ) by translational repression of SOCS1 protein.102 It would be interesting to examine whether different posttranscriptional gene regulatory machineries are employed for the let-7i expression in these two situations during the innate immune response, considering that its miRNA maturation is known to occur under complex control by a network of multiple regulatory factors.
In addition to targeting TLR signaling pathways, multiple lines of evidence have unambiguously indicated that miRNAs can also exert a direct influence on inflammation via inhibition of the production of proinflammatory cytokines or their upstream regulators. In DCs, miR-142-3p was shown to be highly induced after LPS stimulation, resulting in targeted repression of both protein and mRNA levels of IL-6 to suppress inflammation that otherwise causes endotoxin-induced mortality.103 Downregulation of miR-29 was observed in activated natural killer (NK) and T cells from Listeria monocytogenes or Mycobacterium bovis bacillus Calmette-Guérin (BCG)–infected mice, which promotes the production of its mRNA target IFN-γ, contributing to greater host resistance to bacterial infection.104 By directly targeting CaMKII-α (calcium/calmodulin-dependent protein kinase II alpha) for repression, miR-148/152 was shown to inhibit LPS-induced major histocompatibility (MHC) II expression and limit cytokine production in DCs.105 Interestingly, it was reported that excessive inflammation in the brain can be attenuated by induction of miR-132, which targets acetylcholinesterase (AChE), a crucial enzyme hydrolyzing acetylcholine (ACh), which in turn intercepts proinflammatory cytokine production,106 bridging a link between cholinergic signaling and inflammation in neuroimmune disease.






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


