Chapter 3 Electrophysiological Mechanisms of Cardiac Arrhythmias
The mechanisms responsible for cardiac arrhythmias are generally divided into categories of disorders of impulse formation (automaticity or triggered activity), disorders of impulse conduction (reentry), or combinations of both. Automaticity is the property of cardiac cells to initiate an impulse spontaneously, without need for prior stimulation. Triggered activity is impulse initiation in cardiac fibers caused by depolarizing oscillations in membrane voltage (known as afterdepolarizations) that occur consequent to one or more preceding action potentials.1 Reentry occurs when a propagating action potential wave fails to extinguish after initial tissue activation; instead, it blocks in circumscribed areas, circulates around the zones of block, and reenters and reactivates the site of original excitation after it recovers excitability. Reentry is the likely mechanism of most recurrent clinical arrhythmias.
Automaticity
Automaticity, or spontaneous impulse initiation, is the ability of cardiac cells to depolarize spontaneously, reach threshold potential, and initiate a propagated action potential in the absence of external electrical stimulation. Altered automaticity can be caused by enhanced normal automaticity or abnormal automaticity.1
Enhanced Normal Automaticity
Pacemaker Mechanisms
Normal automaticity involves a spontaneous, slow, progressive decline (less negative) in the transmembrane potential during diastole (spontaneous diastolic depolarization or phase 4 depolarization) (see Chap. 1). Once this spontaneous depolarization reaches threshold (approximately −40 mV), a new action potential is generated.2
The ionic mechanisms responsible for normal pacemaker activity in the sinus node are still controversial. The fall in membrane potential during phase 4 seems to arise from a changing balance between positive inward currents, which favor depolarization, and positive outward currents, with a net gain in intracellular positive charges during diastole (i.e., inward depolarizing current; Fig. 3-1).1–6
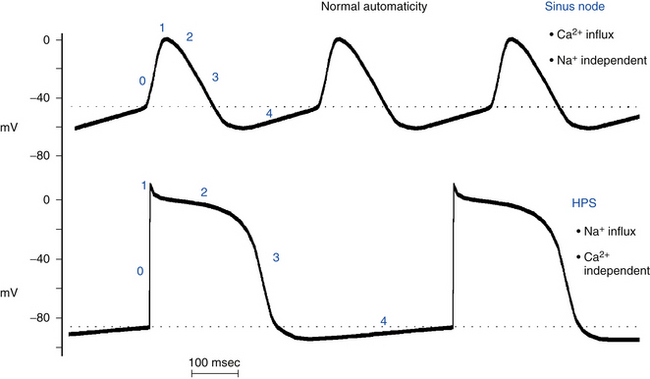
Evidence suggests that If (named the “funny” current because, unlike most voltage-sensitive currents, it is activated by hyperpolarization rather than depolarization) is one of the most important ionic currents involved in the rate regulation of cardiac pacemaker cells, hence its designation as the pacemaker current. If is an inward current carried largely by Na+ and, to a lesser extent, K+ ions. The If channels are deactivated during the action potential upstroke and the initial plateau phase of repolarization. However, they begin to activate at the end of the action potential as repolarization brings the membrane potential to levels more negative than approximately −40 to −50 mV, and If is fully activated at approximately −100 mV. Once activated, If depolarizes the membrane to a level where the Ca2+ current activates to initiate the action potential.7 In its range of activation, which quite properly comprises the voltage range of diastolic depolarization, the current is inward, and its reversal occurs at approximately −10 to −20 mV because of the mixed Na+-K+ permeability of If channels. At the end of the repolarization phase of an action potential, because If activation occurs in the background of a decaying outward (K+ time-dependent) current, current flow quickly shifts from outward to inward, thus giving rise to a sudden reversal of voltage change (from repolarizing to depolarizing) at the maximum diastolic potential. The major role of If has been reinforced by the findings that drugs such as ivabradine targeted to block If slow heart rate and mutations in the If channel are associated with slowed heart rate.2,5,8–10
On the other hand, several studies have shown that If is not the only current that can initiate the diastolic depolarization process in the sinus node. In addition to voltage and time, the electrogenic and regulatory molecules on the surface membrane of sinus node cells are strongly modulated by Ca2+ and phosphorylation, a finding suggesting that intracellular Ca2+ is an important player in controlling pacemaker cell automaticity. Newer evidence points to a substantial impact of another current on the late diastolic depolarization; that is, the Na+-Ca2+ exchanger current activated by submembrane spontaneous rhythmic local Ca2+ releases from the sarcoplasmic reticulum, a major Ca2+ store within sinus node cells, via ryanodine receptors (RyR2). Activation of the local oscillatory Ca2+ releases is independent of membrane depolarization and is driven by a high level of basal state phosphorylation of Ca2+ cycling proteins. Critically timed Ca2+ releases occur during the later phase of diastolic depolarization and activate the forward mode of the Na+-Ca2+ exchanger (one Ca2+ for three Na+). The result is in an inward membrane current that causes the late diastolic depolarization to increase exponentially, thus driving the membrane potential to the threshold to activate a sufficient number of voltage-gated L-type Ca2+ channels and leading to generation of the rapid upstroke of the next action potential (see Fig. 1-3).Although regulated by membrane potential and submembrane Ca2+, the Na+-Ca2+ exchanger does not have time-dependent gating, as do ion channels, but generates an inward current almost instantaneously when submembrane Ca2+ concentration increases.8,9,11
There is some degree of uncertainty about the relative role of If versus that of intracellular Ca2+ cycling in controlling the normal pacemaker cell automaticity and their individual (or mutual) relevance in mediating the positive-negative chronotropic effect of neurotransmitters. Furthermore, the interactions between the membrane ion channel clock and the intracellular Ca2+ clock and the cellular mechanisms underlying this internal Ca2+ clock are not completely elucidated.8,9,11
Hierarchy of Pacemaker Function
Automaticity is not limited to the cells within the sinus node. Under physiological conditions, cells in parts of the atria and within the atrioventricular node (AVN) and the His-Purkinje system (HPS) also possess pacemaking capability. However, the occurrence of spontaneous activity in these cells is prevented by the natural hierarchy of pacemaker function that causes these sites to be latent or subsidiary pacemakers.1 The spontaneous discharge rate of the sinus node normally exceeds that of all other subsidiary pacemakers (see Fig. 3-1). Therefore, the impulse initiated by the sinus node depolarizes subsidiary pacemaker sites and keeps their activity depressed before they can spontaneously reach threshold. However, slowly depolarizing and previously suppressed pacemakers in the atrium, AVN, or ventricle can become active and assume pacemaker control of the cardiac rhythm if the sinus node pacemaker becomes slow or unable to generate an impulse (e.g., secondary to depressed sinus node automaticity) or if impulses generated by the sinus node are unable to activate the subsidiary pacemaker sites (e.g., sinoatrial exit block, or atrioventricular [AV] block). The emergence of subsidiary or latent pacemakers under such circumstances is an appropriate fail-safe mechanism, which ensures that ventricular activation is maintained. Because spontaneous diastolic depolarization is a normal property, the automaticity generated by these cells is classified as normal.
There is also a natural hierarchy of intrinsic rates of subsidiary pacemakers that have normal automaticity, with atrial pacemakers having faster intrinsic rates than AV junctional pacemakers, and AV junctional pacemakers having faster rates than ventricular pacemakers.
Subsidiary Pacemakers
Subsidiary Atrial Pacemakers
Subsidiary atrial pacemakers have been identified in the atrial myocardium, especially in the crista terminalis, at the junction of the inferior right atrium and inferior vena cava, near or on the eustachian ridge, near the coronary sinus ostium, in the atrial muscle that extends into the tricuspid and mitral valves, and in the muscle sleeves that extend into the cardiac veins (venae cavae and pulmonary veins).12
Subsidiary Ventricular Pacemakers
In the ventricles, latent pacemakers are found in the HPS, where Purkinje fibers have the property of spontaneous diastolic depolarization. Isolated cells of the HPS discharge spontaneously at rates of 15 to 60 beats/min, whereas ventricular myocardial cells do not normally exhibit spontaneous diastolic depolarization or automaticity. The relatively slow spontaneous discharge rate of the HPS pacemakers, which further decreases from the His bundle to the distal Purkinje branches, ensures that pacemaker activity in the HPS will be suppressed on a beat-to-beat basis by the more rapid discharge rate of the sinus node and atrial and AV junctional pacemakers. However, enhanced Purkinje fiber automaticity can be induced by certain situations, such as myocardial infarction (MI). In this setting, some Purkinje fibers that survive the infarction develop moderately reduced maximum diastolic membrane potentials and therefore accelerated spontaneous discharge rates.13
Regulation of Pacemaker Function
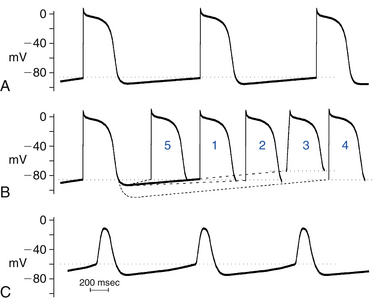
The intrinsic rate at which the sinus node pacemaker cells generate impulses is determined by the interplay of three factors: the maximum diastolic potential, the threshold potential at which the action potential is initiated, and the rate or slope of phase 4 depolarization (Fig. 3-2). A change in any one of these factors will alter the time required for phase 4 depolarization to carry the membrane potential from its maximum diastolic level to threshold and thus alter the rate of impulse initiation.1
The sinus node is innervated by the parasympathetic and sympathetic nervous systems, and the balance between these systems importantly controls the pacemaker rate. The classic concept has been that of a reciprocal relationship between sympathetic and parasympathetic inputs. More recent investigations, however, stress dynamic, demand-oriented interactions, and the anatomical distribution of fibers that allows both autonomic systems to act quite selectively. Muscarinic cholinergic and beta1-adrenergic receptors are nonuniformly distributed in the sinus node, and they modulate both the rate of depolarization and impulse propagation.1
Parasympathetic Activity
Parasympathetic tone reduces the spontaneous discharge rate of the sinus node, whereas its withdrawal accelerates sinus node automaticity. Acetylcholine, the principal neurotransmitter of the parasympathetic nervous system, inhibits spontaneous impulse generation in the sinus node by increasing K+ conductance. Acetylcholine acts through M2 muscarinic receptors to activate the Gi protein, which subsequently results in activation of IKACh (an acetylcholine-activated subtype of inward rectifying current) in tissues of the sinus node and AVN as well as of the atria, Purkinje fibers, and ventricles. The increased outward repolarizing K+ current (IK) leads to membrane hyperpolarization (i.e., the resting potential and the maximum diastolic potential become more negative). The resulting hyperpolarization of the membrane potential lengthens the time required for the membrane potential to depolarize to threshold and thereby decreases the automaticity of the sinus node (see Fig. 3-2). In addition, activation of the Gi protein results in inhibition of beta-receptor–stimulated adenylate cyclase activity, thus reducing cyclic adenosine monophosphate (cAMP) and inhibiting protein kinase A, with subsequent inhibition of the inward Ca2+ current. This results in reduction of the rate of diastolic depolarization because of less Ca2+ entry and subsequent slowing of the pacemaker activity. Inhibition of beta-receptor–stimulated adenylate cyclase activity can also inhibit the inward If current.
Sympathetic Activity
Increased sympathetic nerve traffic and the adrenomedullary release of catecholamines increase sinus node discharge rate. Stimulation of beta1-receptors by catecholamines enhances the L-type of inward Ca2+ current (ICaL) by increasing cAMP and activating the protein kinase A system; the increment in inward Ca2+ current increases the slope of diastolic depolarization and enhances pacemaker activity (see Fig. 3-2). The redistribution of Ca2+ can also increase the completeness and the rate of deactivation of the rapid (IKr) and slow (IKs) components of the delayed rectifier IK; the ensuing decline in the opposing outward current results in a further net increase in inward current. Catecholamines can also enhance the inward If current by shifting the voltage dependence of If to more positive potentials, thus augmenting the slope of phase 4 and increasing the rate of sinus node firing.10
Atrial, AV junctional, and HPS subsidiary pacemakers are also under similar autonomic control, with the sympathetic nervous system enhancing pacemaker activity through beta1-adrenergic stimulation and the parasympathetic nervous system inhibiting pacemaker activity through muscarinic receptor stimulation.1
Other Influences
Evidence indicates that active and passive changes in the mechanical environment of the heart provide feedback to modify cardiac rate and rhythm and are capable of influencing both the initiation and spread of cardiac excitation. This direction of the crosstalk between cardiac electrical and mechanical activity is referred to as mechanoelectric feedback and is thought to be involved in the adjustment of heart rate to changes in mechanical load, which would help explain the precise beat-to-beat regulation of cardiac performance. Acute mechanical stretch enhances automaticity, reversibly depolarizes the cell membrane, and shortens the action potential duration. Feedback from cardiac mechanics to electrical activity involves mechanosensitive ion channels and ATP-sensitive K+ channels. In addition, Na+ and Ca2+ entering the cells via nonselective ion channels are thought to contribute to the genesis of stretch-induced arrhythmia.14
Abnormal Automaticity
In the normal heart, automaticity is confined to the sinus node and other specialized conducting tissues. Working atrial and ventricular myocardial cells do not normally have spontaneous diastolic depolarization and do not initiate spontaneous impulses, even when they are not excited for long periods of time by propagating impulses. Although these cells do have an If, the range of activation of this current in these cells is much more negative (−120 to −170 mV) than in Purkinje fibers or in the sinus node. As a result, during physiological resting membrane potentials (−85 to −95 mV), the If is not activated, and ventricular cells do not depolarize spontaneously.1 When the resting potentials of these cells are depolarized sufficiently, to approximately −70 to −30 mV, however, spontaneous diastolic depolarization can occur and cause repetitive impulse initiation, a phenomenon called depolarization-induced automaticity or abnormal automaticity (see Fig. 3-2). Similarly, cells in the Purkinje system, which are normally automatic at high levels of membrane potential, show abnormal automaticity when the membrane potential is reduced to approximately −60 mV or less, as can occur in ischemic regions of the heart. When the steady-state membrane potential of Purkinje fibers is reduced to approximately −60 mV or less, the If channels that participate in normal pacemaker activity in Purkinje fibers are closed and nonfunctional, and automaticity is therefore not caused by the normal pacemaker mechanism. It can, however, be caused by an “abnormal” mechanism. In contrast, enhanced automaticity of the sinus node, subsidiary atrial pacemakers, or the AVN caused by a mechanism other than acceleration of normal automaticity has not been demonstrated clinically.15
Several different mechanisms probably cause abnormal pacemaker activity at low membrane potentials, including activation and deactivation of the delayed rectifier IK, intracellular Ca2+ release from the sarcoplasmic reticulum that causes activation of inward Ca2+ currents and the inward INa (through the Na+-Ca2+ exchanger), and a potential contribution by If.16 It has not been determined which of these mechanisms are operative in the different pathological conditions in which abnormal automaticity can occur.1
The upstroke of the spontaneously occurring action potentials generated by abnormal automaticity can be caused by Na+ or Ca2+ inward currents or possibly a combination of the two.1 In the range of diastolic potentials between approximately −70 and −50 mV, repetitive activity is dependent on extracellular Na+ concentration and can be decreased or abolished by Na+ channel blockers. In a diastolic potential range of approximately −50 to −30 mV, Na+ channels are predominantly inactivated; repetitive activity depends on extracellular Ca2+ concentration and is reduced by L-type Ca2+ channel blockers.
The intrinsic rate of a focus with abnormal automaticity is a function of the membrane potential. The more positive the membrane potential is, the faster the automatic rate will be (see Fig. 3-2). Abnormal automaticity is less vulnerable to suppression by overdrive pacing (see later). Therefore, even occasional slowing of the sinus node rate can allow an ectopic focus with abnormal automaticity to fire without a preceding long period of quiescence. Catecholamines can increase the rate of discharge caused by abnormal automaticity and therefore can contribute to a shift in the pacemaker site from the sinus node to a region with abnormal automaticity.
Overdrive Suppression of Automatic Rhythms
Suppression of Normal and Abnormal Automatic Subsidiary Pacemakers
The sinus node likely maintains its dominance over subsidiary pacemakers in the AVN and the Purkinje fibers by several mechanisms. During sinus rhythm in a normal heart, the intrinsic automatic rate of the sinus node is faster than that of the other potentially automatic cells. Consequently, the latent pacemakers are excited by propagated impulses from the sinus node before they have a chance to depolarize spontaneously to threshold potential. The higher frequency of sinus node discharge also suppresses the automaticity of other pacemaker sites by a mechanism called overdrive suppression. The diastolic (phase 4) depolarization of the latent pacemaker cells with the property of normal automaticity is actually inhibited because the cells are repeatedly depolarized by the impulses from the sinus node.1 Electrotonic interaction between the pacemaker cells and the nonpacemaker cells in the surrounding myocardium via intercalated discs and gap junctions can also hyperpolarize the latent pacemakers and contribute to their suppression (Fig. 3-3).
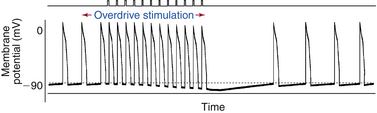
Mechanism of Overdrive Suppression
The mechanism of overdrive suppression is mediated mostly by enhanced activity of the Na+-K+ exchange pump that results from driving a pacemaker cell faster than its intrinsic spontaneous rate. During normal sinus rhythm (NSR), latent pacemakers are depolarized at a higher frequency than their intrinsic rate of automaticity. The increased frequency of depolarizations leads to an increase in intracellular Na+, which enters the cell with every action potential, because more Na+ enters the cell per unit time.1 The increased intracellular Na+ stimulates the Na+-K+ exchange pump. Because the Na+-K+ exchange pump is electrogenic (i.e., moves more Na+ outward than K+ inward), it generates a net outward (hyperpolarizing) current across the cell membrane. This drives the membrane potential more negative, thereby offsetting the depolarizing If being carried into the cell and slowing the rate of phase 4 diastolic depolarization. This effectively prevents the If from depolarizing the cell to its threshold potential and thereby suppresses spontaneous impulse initiation in these cells.
Abnormally automatic cells and tissues at reduced levels of membrane potential are less sensitive to overdrive suppression than cells and tissues that are fully polarized, with enhanced normal automaticity. The amount of overdrive suppression of spontaneous diastolic depolarization that causes abnormal automaticity is directly related to the level of membrane potential at which the automatic rhythm occurs. At low levels of membrane potential, Na+ channels are inactivated, decreasing the fast inward INa; therefore, there are reductions in the amount of Na+ entering the cell during overdrive and the degree of stimulation of the Na+-K+ exchange pump. The more polarized the membrane is during phase 4, the larger the amount will be of Na+ entering the cell with each action potential, and the more overdrive suppression will occur. As a result of the lack of overdrive suppression of abnormally automatic cells, even transient sinus pauses can permit an ectopic focus with a slower rate than the sinus node to capture the heart for one or more beats. However, even in situations in which the cells can be sufficiently depolarized to inactivate the INa and limit intracellular Na+ load, overdrive suppression can still be observed because of increased intracellular Ca2+ loading. Such Ca2+ loading can activate Ca2+-dependent K+ conductance (favoring repolarization) and promote Ca2+ extrusion through the Na+-Ca2+ exchanger and Ca2+ channel phosphorylation, thus increasing Na+ load and thus Na+-K+ exchange pump activity. The increase in intracellular Ca2+ load can also reduce the depolarizing ICaL by promoting Ca2+-induced inactivation of the Ca2+ current.
Arrhythmias Caused by Automaticity
Parasystole
Parasystole is a result of interaction between two fixed rate pacemakers having different discharge rates. Parasystolic pacemakers can exist in either the atrium or the ventricle. The latent pacemaker is protected from being overdriven by the dominant rhythm (usually NSR) by intermittent or constant entrance block (i.e., impulses of sinus origin fail to depolarize the latent pacemaker secondary to block in the tissue surrounding the latent pacemaker focus). Various mechanisms have been postulated to explain the protected zone surrounding the ectopic focus. It is possible that the depolarized level of membrane potential at which abnormal automaticity occurs can cause entrance block, leading to parasystole. This would be an example of an arrhythmia caused by a combination of an abnormality of impulse conduction and impulse initiation. Such block, however, must be unidirectional, so that activity from the ectopic pacemaker can exit and produce depolarization whenever the surrounding myocardium is excitable. The protected pacemaker is said to be a parasystolic focus. In general, under these conditions, a protected focus of automaticity of this type fires at its own intrinsic frequency, and the intervals between the discharges of each pacemaker are multiples of its intrinsic discharge rate (sometimes described as fixed parasystole). Therefore, on the surface ECG, the coupling intervals of the manifest ectopic beats wander through the basic cycle of the sinus rhythm. Accordingly, the traditional ECG criteria used to recognize the fixed form of parasystole are (1) the presence of variable coupling intervals of the manifest ectopic beats, (2) interectopic intervals that are simple multiples of a common denominator, and (3) the presence of fusion beats. Occasionally, the parasystolic focus can exhibit exit block, during which it may fail to depolarize excitable myocardium.17
Although the parasystolic focus is protected, it may not be totally immune to the surrounding electrical activity. The effective electrical communication that permits the emergence of the ectopic discharges can also allow the rhythmic activity of the surrounding tissues to electrotonically influence the periodicity of the pacemaker discharge rate (described as modulated parasystole). Electrotonic influences arriving during the early stage of diastolic depolarization result in a delay in the firing of the parasystolic focus, whereas those arriving late accelerate the discharge of the parasystolic focus. As a consequence, the dominant pacemaker can entrain the partially protected parasystolic focus and force it to discharge at periods that may be faster or slower than its own intrinsic cycle and give rise to premature discharges whose patterns depend on the degree of modulation and the basic heart rate, occasionally mimic reentry, and occur at fixed coupling intervals. Therefore, appropriate diagnosis of modulated parasystole relies on the construction of a phase response curve as theoretical evidence of modulation of the ectopic pacemaker cycle length (CL) by the electrotonic activity generated by the sinus discharges across the area of protection.17
Arrhythmias Caused by Abnormal Automaticity
There appears to be an association between abnormal Purkinje fiber automaticity and the arrhythmias that occur during the acute phase of MI (e.g., an accelerated idioventricular rhythm). However, the role of abnormal automaticity in the development of ventricular arrhythmias associated with chronic ischemic heart disease is less certain. Additionally, isolated myocytes obtained from hypertrophied and failing hearts have been shown to manifest spontaneous diastolic depolarization and enhanced If, findings suggesting that abnormal automaticity can contribute to the occurrence of some arrhythmias in heart failure and left ventricular hypertrophy.
Triggered Activity
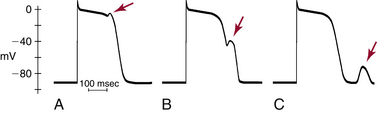
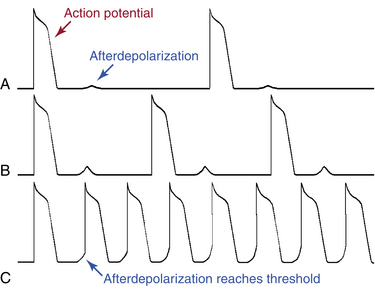
Triggered activity is impulse initiation in cardiac fibers caused by afterdepolarizations that occur consequent to a preceding impulse or series of impulses.1 Afterdepolarizations are depolarizing oscillations in membrane potential that follow the upstroke of a preceding action potential. Afterdepolarizations can occur early during the repolarization phase of the action potential (early afterdepolarization [EAD]) or late, after completion of the repolarization phase (delayed afterdepolarization [DAD]; Fig. 3-4). When either type of afterdepolarization is large enough to reach the threshold potential for activation of a regenerative inward current, a new action potential is generated, which is referred to as triggered.
Delayed Afterdepolarizations and Triggered Activity
DADs are oscillations in membrane voltage that occur after completion of repolarization of the action potential (i.e., during phase 4). The transient nature of the DAD distinguishes it from normal spontaneous diastolic (pacemaker) depolarization, during which the membrane potential declines almost monotonically until the next action potential occurs. DADs may or may not reach threshold. Subthreshold DADs do not initiate action potentials or trigger arrhythmias. When a DAD does reach threshold, only one triggered action potential occurs (Fig. 3-5). The triggered action potential can also be followed by a DAD that, again, may or may not reach threshold and may or may not trigger another action potential. The first triggered action potential is often followed by a short or long train of additional triggered action potentials, each arising from the DAD caused by the previous action potential.
Ionic Basis of Delayed Afterdepolarizations
DADs usually occur under a variety of conditions in which Ca2+ overload develops in the cytoplasm and sarcoplasmic reticulum. During the plateau phase of the normal action potential, Ca2+ flows through voltage-dependent L-type Ca2+ channels (ICaL). Although the rise in intracellular Ca2+ is small and not sufficient to induce contraction, the small amount of Ca2+ entering the cell via ICaL triggers a massive release of Ca2+ from the sarcoplasmic reticulum (the major store for Ca2+) into the cytosol by opening the RyR2 channels (present in the membrane of the sarcoplasmic reticulum) in a process known as Ca2+-induced Ca2+ release (CICR).1,6 During repolarization (i.e., diastole), most of the surplus Ca2+ in the cytosol is resequestered into the sarcoplasmic reticulum by the sarcoplasmic reticulum Ca2+ adenosine triphosphatase (SERCA), the activity of which is controlled by the phosphoprotein phospholamban. Additionally, some of the Ca2+ is extruded from the cell by the Na+-Ca2+ exchanger to balance the Ca2+ that enters with Ca2+ current. Recurring Ca2+ release-uptake cycles provide the basis for periodic elevations of the cytosolic Ca2+ concentration and contractions of myocytes, hence for the orderly beating of the heart (Fig. 3-6).18–20
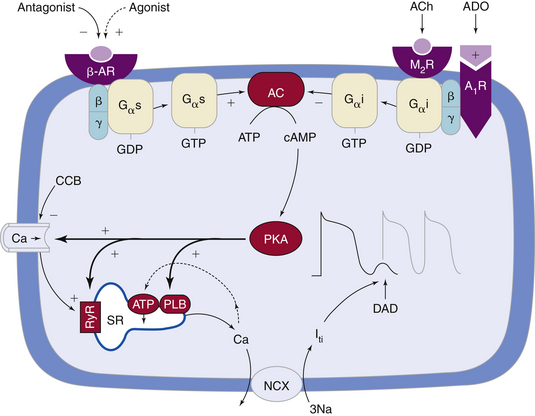
Under various pathological conditions, Ca2+ concentration in the sarcoplasmic reticulum can rise to a critical level during repolarization (i.e., Ca2+ overload), at which time a secondary spontaneous release of Ca2+ from the sarcoplasmic reticulum occurs after the action potential, rather than as a part of excitation-contraction coupling. This secondary release of Ca2+ results in inappropriately timed Ca2+ transients and contractions. Spontaneous Ca2+ waves are arrhythmogenic; they induce Ca2+-dependent depolarizing membrane currents (transient inward current), mainly by activation of the Na+-Ca2+ exchanger, thereby causing oscillations of the membrane potential known as DADs. After one or several DADs, myoplasmic Ca2+ can decrease because the Na+-Ca2+ exchanger extrudes Ca2+ from the cell, and the membrane potential stops oscillating.18–20
When the DADs are of low amplitude, they usually are not apparent or clinically significant. However, during pathological conditions (e.g., myocardial ischemia, acidosis, hypomagnesemia, digitalis toxicity, and increased catecholamines), the amplitude of the Ca2+-mediated oscillations is increased and can reach the stimulation threshold, and an action potential is triggered. If this process continues, sustained tachycardia will develop. Probably the most important influence that causes subthreshold DADs to reach threshold is a decrease in the initiating CL, because that increases both the amplitude and rate of the DADs. Therefore, initiation of arrhythmias triggered by DADs can be facilitated by a spontaneous or pacing-induced increase in the heart rate.
Digitalis causes DAD-dependent triggered arrhythmias by inhibiting the Na+-K+ exchange pump. In toxic amounts, this effect results in the accumulation of intracellular Na+ and, consequentially, an enhancement of the Na+-Ca2+ exchanger in the reverse mode (Na+ removal, Ca2+ entry) and an accumulation of intracellular Ca2+.16 Spontaneously occurring accelerated ventricular arrhythmias that occur during digitalis toxicity are likely to be caused by DADs. Triggered ventricular arrhythmias caused by digitalis also can be initiated by pacing at rapid rates. As toxicity progresses, the duration of the trains of repetitive responses induced by pacing increases.
Catecholamines can cause DADs by increasing intracellular Ca2+ overload secondary to different mechanisms. Catecholamines increase the slow, inward ICaL through stimulation of beta-adrenergic receptors and increasing cAMP, which result in an increase in transsarcolemmal Ca2+ influx and intracellular Ca2+ overload (see Fig. 3-6). Catecholamines can also enhance the activity of the Na+-Ca2+ exchanger, thus increasing the likelihood of DAD-mediated triggered activity. Additionally, catecholamines enhance the uptake of Ca2+ by the sarcoplasmic reticulum and lead to increased Ca2+ stored in the sarcoplasmic reticulum and the subsequent release of an increased amount of Ca2+ from the sarcoplasmic reticulum during contraction.16 Sympathetic stimulation can potentially cause triggered atrial and ventricular arrhythmias, possibly some of the ventricular arrhythmias that accompany exercise and those occurring during ischemia and infarction.
Elevations in intracellular Ca2+ in the ischemic myocardium are also associated with DADs and triggered arrhythmias. Accumulation of lysophosphoglycerides in the ischemic myocardium, with consequent Na+ and Ca2+ overload, has been suggested as a mechanism for DADs and triggered activity. Cells from damaged areas or surviving the infarction can display spontaneous release of Ca2+ from sarcoplasmic reticulum, which can generate waves of intracellular Ca2+ elevation and arrhythmias.19,20
Abnormal sarcoplasmic reticulum function caused by genetic defects that impair the ability of the sarcoplasmic reticulum to sequester Ca2+ during diastole can lead to DADs and be the cause of certain inherited ventricular tachyarrhythmias. Mutations in the cardiac RyR2, the sarcoplasmic reticulum Ca2+ release channel in the heart, have been identified in kindreds with the syndrome of catecholaminergic polymorphic VT and ventricular fibrillation (VF) with short QT intervals. It seems likely that perturbed intracellular Ca2+, and perhaps also DADs, underlie arrhythmias in this syndrome (see Fig. 3-6).18,21
Several drugs can inhibit DAD-related triggered activity via different mechanisms, including reduction of the inward Ca2+ current and intracellular Ca2+ overload (Ca2+ channel blockers, beta-adrenergic blockers; see Fig. 3-6), reduction of Ca2+ release from the sarcoplasmic reticulum (caffeine, ryanodine, thapsigargin, cyclopiazonic acid), and reduction of the inward INa (tetrodotoxin, lidocaine, phenytoin).
DAD-related triggered activity is thought to be a mechanism for tachyarrhythmia associated with MI, reperfusion injury, some right ventricular outflow tract tachycardia, and some atrial tachyarrhythmias. DADs are more likely to occur with fast spontaneous or paced rates or with increased premature beats.15,18,20,22
Properties of Delayed Afterdepolarizations
The amplitude of DADs and the coupling interval between the first triggered impulse and the last stimulated impulse that induced them are directly related to the drive CL at which triggered impulses are initiated. A decrease in the basic drive CL (even a single drive cycle; i.e., premature impulse), in addition to increasing the DAD amplitude, results in a decrease in the coupling interval between the last drive cycle and the first DAD-triggered impulse, with respect to the last driven action potential, and an increase of the rate of DADs. Triggered activity tends to be induced by a critical decrease in the drive CL, either spontaneous, such as in sinus tachycardia, or pacing induced. The increased time during which the membrane is in the depolarized state at shorter stimulation CLs or after premature impulses increases Ca2+ in the myoplasm and the sarcoplasmic reticulum, thus increasing the transient inward current responsible for the increased afterdepolarization amplitude, causing the current to reach its maximum amplitude more rapidly, and decreasing the coupling interval of triggered impulses. The repetitive depolarizations can increase intracellular Ca2+ because of repeated activation of ICaL. This characteristic property can help distinguish triggered activity from reentrant activity because the relationship for reentry impulses initiated by rapid stimulation is often the opposite; that is, as the drive CL is reduced, the first reentrant impulse occurs later with respect to the last driven action potential because of rate-dependent conduction slowing in the reentrant pathway.
Early Afterdepolarizations and Triggered Activity
Ionic Basis of Early Afterdepolarizations
The plateau of the action potential is a time of high membrane resistance (i.e., membrane conductance to all ions falls to rather low values), when there is little current flow. Consequently, small changes in repolarizing or depolarizing currents can have profound effects on the action potential duration and profile. Normally, during phases 2 and 3, the net membrane current is outward. Any factor that transiently shifts the net current in the inward direction can potentially overcome and reverse repolarization and lead to EADs. Such a shift can arise from blockage of the outward current, carried by Na+ or Ca2+ at that time, or enhancement of the inward current, mostly carried by K+ at that time.1
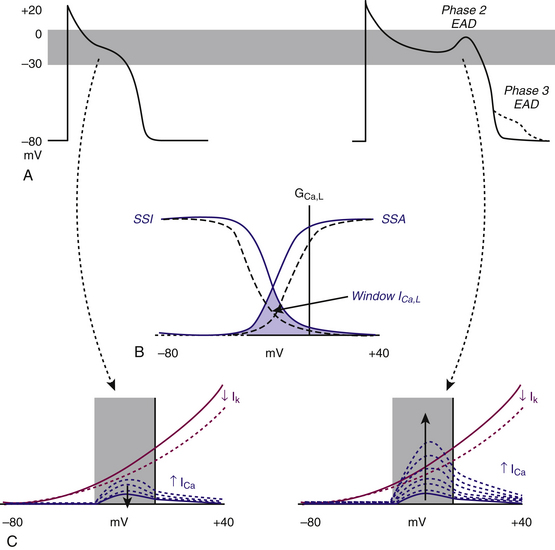
EADs have been classified as phase 2 (occurring at the plateau level of membrane potential) and phase 3 (occurring during phase 3 of repolarization; see Fig. 3-4). The ionic mechanisms of phase 2 and phase 3 EADs and the upstrokes of the action potentials they elicit can differ.1 At the depolarized membrane voltages of phase 2, Na+ channels are inactivated; hence, the ICaL and Na+-Ca2+ exchanger current are the major currents potentially responsible for EADs. Voltage steady-state activation and inactivation of the L-type Ca2+ channels are sigmoidal, with an activation range over –40 to +10 mV (with a half-activation potential near –15 mV) and a half-inactivation potential near –35 mV. However, a relief of inactivation for voltages positive to 0 mV leads to a U-shaped voltage curve for steady-state inactivation. Overlap of the steady-state voltage-dependent inactivation and activation relations defines a “window” current near the action potential plateau, within which transitions from closed and open states can occur. As the action potential repolarizes into the window region, ICaL increases and can potentially be sufficient to reverse repolarization, thus generating the EAD upstroke (Fig. 3-7).23
< div class='tao-gold-member'>






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


