Chapter 8 Sinus Node Dysfunction
General Considerations
Anatomy and Physiology of the Sinus Node
The sinus node is the dominant pacemaker of the heart. Its pacemaker function is determined by its low maximum diastolic membrane potential and steep phase 4 spontaneous depolarization. The molecular mechanisms of pacemaker function of the sinus node are discussed in detail in Chapters 1 and 3.1
The sinus node is a subepicardial specialized muscular structure located laterally within the epicardial groove of the sulcus terminalis of the right atrium (RA) at the junction of the anterior trabeculated appendage with the posterior smooth-walled venous component. The endocardial aspect of the sulcus terminalis is marked by the crista terminalis. Starting epicardially at the junction of the superior vena cava (SVC) and the RA appendage, it courses downward and to the left along the sulcus terminalis, to end subendocardially almost to the inferior vena cava (IVC). The sinus node is a spindle-shaped structure with a central body and tapering ends; the head extends toward the interatrial groove, and the tail extends toward the orifice of IVC. In adults, the sinus node measures 10 to 20 mm long and 2 to 3 mm wide and thick.1–5
The sinus node consists of densely packed specialized myocytes of no definite orientation within a background of extracellular connective tissue matrix.4 The nodal margins can be discrete with fibrous separation from the surrounding atrial myocardium or interdigitate though a transitional zone. Commonly, prongs of nodal (P) cells and transitional (T) cells extend from the nodal body into the atrial myocardium, but actual cell-to-cell interaction is uncertain.6
The pacemaker activity is not confined to a single cell in the sinus node; rather, sinus node cells function as electrically coupled oscillators that discharge synchronously because of mutual entrainment. In fact, it is likely that sinus rhythm results from impulse origin at widely separated sites, with two or three individual wavefronts created that merge to form a single, widely disseminated wavefront. The sinus node is insulated electrically from the surrounding atrial myocytes, except at a limited number of preferential exit sites. Neural and hormonal factors influence both the site of pacemaker activation, likely via shifting points of initial activity, and the point of exit from the sinus node complex. At faster rates, the sinus impulse originates in the superior portion of the sinus node, whereas at slower rates, it arises from a more inferior portion.7 High-density simultaneous endocardial unipolar mapping studies demonstrated the frequent occurrence of spontaneous variations in the P wave and sinus activation sequence in normal individuals. These findings suggested that the sinus node complex in normal hearts displays a dynamic range of activation sites along the posterolateral RA. Furthermore, preferential pathways of conduction were also found to exist between the sinus node and the atrial exit sites, thus potentially contributing to the multicentricity of the sinus node complex.1,5,8
The blood supply to the sinus node region is variable and is therefore vulnerable to damage during operative procedures. The blood supply predominantly comes from a large central artery, the sinus nodal artery, which is a branch of the right coronary artery in 55% to 60% of patients, and from the circumflex artery in 40% to 45%. The sinus nodal artery typically passes centrally through the length of the sinus body, and it is disproportionately large, which is considered physiologically important in that its perfusion pressure can affect the sinus rate. Distention of the artery slows the sinus rate, whereas collapse causes an increase in rate.1,3
The sinus node is densely innervated with postganglionic adrenergic and cholinergic nerve terminals (threefold greater density of beta-adrenergic and muscarinic cholinergic receptors than adjacent atrial tissue), both of which influence the rate of spontaneous depolarization in pacemaker cells and can cause a shift in the principal pacemaker site within the sinus node region, which is often associated with subtle changes in P wave morphology. Enhanced vagal activity can produce sinus bradycardia, sinus arrest, and sinoatrial exit block, whereas increased sympathetic activity can increase the sinus rate and reverse sinus arrest and sinoatrial exit block. Sinus node responses to brief vagal bursts begin after a short latency and dissipate quickly; in contrast, responses to sympathetic stimulation begin and dissipate slowly. The rapid onset and offset of responses to vagal stimulation allow dynamic beat-to-beat vagal modulation of the heart rate, whereas the slow temporal response to sympathetic stimulation precludes any beat-to-beat regulation by sympathetic activity.1
Periodic vagal bursting (as may occur each time a systolic pressure wave arrives at the baroreceptor regions in the aortic and carotid sinuses) induces phasic changes in the sinus cycle length (CL) and can entrain the sinus node to discharge faster or slower at periods identical to those of the vagal burst.3 Because the peak vagal effects on sinus rate and atrioventricular node (AVN) conduction occur at different times in the cardiac cycle, a brief vagal burst can slow the sinus rate without affecting AVN conduction or can prolong AVN conduction time and not slow the sinus rate.1
Pathophysiology of Sinus Node Dysfunction
The cause of sinus node dysfunction (SND) can be classified as intrinsic (secondary to a pathological condition involving the sinus node proper) or extrinsic (caused by depression of sinus node function by external factors such as drugs or autonomic influences).
Intrinsic Sinus Node Dysfunction
Idiopathic degenerative disease is probably the most common cause of intrinsic SND.2 Ischemic heart disease can be responsible for one third of cases of SND. Transient slowing of the sinus rate or sinus arrest can complicate acute myocardial infarction, which is usually seen with acute inferior wall infarction and is caused by autonomic influences. Possible mechanisms for sinus bradycardia after an acute myocardial infarction include neurological reflexes (Bezold-Jarisch reflex), coronary chemoreflexes (vagally mediated), humoral reflexes (enzymes, adenosine, potassium [K+]), oxygen-conserving reflex (“diving” reflex), and infarction or ischemia of the sinus node or the surrounding atrium (e. g., secondary to proximal occlusion of the right or the circumflex coronary artery).
Cardiomyopathy, long-standing hypertension, infiltrative disorders (e.g., amyloidosis and sarcoidosis), collagen vascular diseases, and surgical trauma can also result in SND.9,10 Orthotropic cardiac transplantation with atrial-atrial anastomosis is associated with a high incidence of SND in the donor heart (likely because of sinus nodal artery damage). Musculoskeletal disorders such as myotonic dystrophy or Friedreich ataxia are rare causes of SND. Congenital heart disease, such as sinus venosus and secundum atrial septal defects, can be associated with SND, even though no surgery has been performed.11 Surgical trauma is responsible for most cases of SND in the pediatric population. Most commonly associated with this complication is the Mustard procedure for transposition of the great arteries and repair of atrial septal defects, especially of the sinus venosus type.11
Furthermore, atrial tachyarrhythmias can precipitate SND, likely secondary to remodeling of sinus node function. Although early studies implicated anatomical structural abnormalities in the sinus node, which suggested a fixed SND substrate, more recent evidence implicated a functional, and potentially reversible, component involving remodeling of sinus node ion channel expression and function. This finding was supported clinically by the observation that successful catheter ablation of atrial fibrillation (AF) and atrial flutter can be followed by significant improvements in sinus node function. In particular, downregulation of the funny current (If) and malfunction of the calcium (Ca2+) clock (characterized by reduced sarcoplasmic reticulum Ca2+ release and downregulated ryanodine receptors in the sinus node) seem to account largely for atrial tachycardia–induced remodeling of sinus node. The remodeled atria are associated with more caudal activation of the sinus node complex, slower conduction time along preferential pathways, and only modest shifts within the functional pacemaker complex.5,12,13
On the other hand, SND has been associated with an increased propensity of atrial tachyarrhythmias, AF in particular. The mechanism leading to AF in patients with SND is unlikely to be bradycardia-dependent because AF was found to develop despite pacing in these patients. Importantly, patients with SND appear to have more widespread atrial changes beyond the sinus node, a finding indicating atrial myopathy, as evidenced by increased atrial refractoriness, prolonged P wave duration, delayed conduction, slowing electrogram fractionation, regions of low voltage and scar, and caudal shift of the pacemaker complex with loss of normal multicentric pattern of activation. Furthermore, abnormal atrial electromechanical properties, chronic atrial stretch, and neurohormonal activation are likely contributors to SND and its related atrial myopathy. The diffuse atrial myopathy potentially underlies the increased propensity to AF. The cause of these diffuse atrial abnormalities remains unknown, but there appears to be a relationship between atrial remodeling that predisposes to AF and sinus node remodeling that results in SND.5,14
Genetic defects in ion channels and structural proteins have been shown to contribute to SND, manifesting as sinus bradycardia, sinus arrest, sinoatrial block, or a combination. Mutations in the SCN5A gene (which encodes the alpha subunit of the cardiac sodium [Na+] channel [INa]), the HCN4 gene (which encodes the protein that contributes to formation of If channels), the KCNQ1 gene (which encodes the alpha subunit of the voltage-gated slowly activating delayed rectifier K+ channel responsible for IKs), the GJA5 gene (which encodes for connexin 40, a gap junction protein), the ANK2 gene (which encodes for ankyrin, which links the integral membrane proteins to the underlying cytoskeleton), and the EMD gene (which encodes the nuclear membrane protein emerin) have been associated with familial forms of SND, many of which also exhibit an increased propensity to AF.15–18
Extrinsic Sinus Node Dysfunction
In the absence of structural abnormalities, the predominant causes of SND are drug effects and autonomic influences. Drugs can alter sinus node function by direct pharmacological effects on nodal tissue or indirectly by neurally mediated effects.19 Drugs known to depress sinus node function include beta blockers, calcium channel blockers (verapamil and diltiazem), digoxin, sympatholytic antihypertensive agents (e.g., clonidine), and antiarrhythmic agents (classes IA, IC, and III).
SND can sometimes result from excessive vagal tone in individuals without intrinsic sinus node disease. Hypervagotonia can be seen in hypersensitive carotid sinus syndrome and neurocardiogenic syncope. Well-trained athletes with increased vagal tone occasionally may require some deconditioning to help prevent symptomatic bradyarrhythmias.20 Surges in vagal tone also can occur during Valsalva maneuvers, endotracheal intubation, vomiting, and suctioning. Sinus slowing in this setting is characteristically paroxysmal and may be associated with evidence of AV conduction delay, secondary to effects of the enhanced vagal tone on both the sinus node and AVN. Less common extrinsic causes of SND include electrolyte abnormalities such as hyperkalemia, hypothermia, increased intracranial pressure (the Cushing response), sleep apnea, hypoxia, hypercapnia, hypothyroidism, advanced liver disease, typhoid fever, brucellosis, and sepsis.
Clinical Presentation
More than 50% of the patients with SND are older than 50 years. Patients often are asymptomatic or have symptoms that are mild and nonspecific, and the intermittent nature of these symptoms makes documentation of the associated arrhythmia difficult at times. Symptoms, which may have been present for months or years, include paroxysmal dizziness, presyncope, or syncope, which are predominantly related to prolonged sinus pauses. Episodes of syncope are often unheralded and can manifest in older patients as repeated falls. The highest incidence of syncope associated with SND probably occurs in patients with tachycardia-bradycardia syndrome, in whom syncope typically occurs secondary to a long sinus pause following cessation of the supraventricular tachycardia (usually AF). Occasionally, a stroke can be the first manifestation of SND in patients presenting with paroxysmal AF and thromboembolism.3,19,21
Patients with sinus bradycardia or chronotropic incompetence can present with decreased exercise capacity or fatigue (Fig. 8-1). Chronotropic incompetence is estimated to be present in 20% to 60% of patients with SND.22,23 Other symptoms include irritability, nocturnal wakefulness, memory loss, lightheadedness, and lethargy. More subtle symptoms include mild digestive disturbances, periodic oliguria or edema, and mild intermittent dyspnea. Additionally, symptoms caused by the worsening of conditions such as congestive heart failure and angina pectoris can be precipitated by SND.
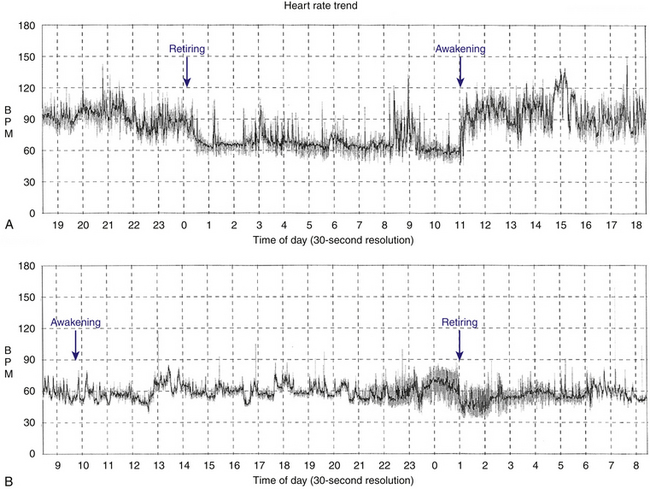
Natural History
The natural history of SND can be variable, but slow progression (over 10 to 30 years) is expected. The prognosis largely depends on the type of dysfunction and the presence and severity of the underlying heart disease. The worst prognosis is associated with the tachycardia-bradycardia syndrome (mostly because of the risk for thromboembolic complications), whereas sinus bradycardia is much more benign. The incidence of new-onset AF in patients with SND is about 5.2% per year. New atrial tachyarrhythmias occur with less frequency in patients who are treated with atrial pacing (3.9%) compared with a greatly increased incidence of similar arrhythmias in patients with only ventricular pacing (22.3%).24 Furthermore, thromboembolism occurs in 15.2% among unpaced patients with SND versus 13% among patients treated with only ventricular pacing versus 1.6% among those treated with atrial pacing.19,21,25
The incidence of advanced AV conduction system disease in patients with SND is low (5% to 10%), and, when present, its progression is slow. At the time of diagnosis of SND, approximately 17% of the patients have some degree of AV conduction system disease (PR interval longer than 240 milliseconds, bundle branch block, His bundle–ventricular (HV) interval prolongation, AV Wenckebach rate less than 120 beats/min, or second- or third-degree AV block). New AV conduction abnormalities develop at a rate of approximately 2.7% per year. The incidence of advanced AV block during long-term follow-up is low (approximately 1% per year).21
Diagnostic Evaluation
Generally, the noninvasive methods of ECG monitoring, exercise testing, and autonomic testing are used first. However, if symptoms are infrequent and noninvasive evaluation is unrevealing, invasive electrophysiological (EP) testing may be pursued.26
Electrocardiogram and Ambulatory Monitoring
A 12-lead electrocardiogram (ECG) needs to be obtained in symptomatic patients. However, the diagnosis of SND as the cause of the symptoms is rarely made from the ECG. In patients with frequent symptoms, 24- or 48-hour ambulatory Holter monitoring can be useful. Cardiac event monitoring or implantable loop recorders may be necessary in patients with less frequent symptoms.27 Documentation of symptoms in a diary by the patient while wearing the cardiac monitor is essential for correlation of symptoms with the heart rhythm at the time. In some cases, ambulatory monitoring can exclude SND as the cause of symptoms if normal sinus rhythm (NSR) is documented at the time of symptom occurrence. In contrast, recorded sinus pauses may not be associated with symptoms.
Autonomic Modulation
An abnormal response to carotid sinus massage (pause longer than 3 seconds) can indicate SND, but this response can also occur in asymptomatic older individuals. Heart rate response to the Valsalva maneuver (normally decreased) or upright tilt (normally increased) can also be used to verify that the autonomic nervous system itself is intact. Complete pharmacological autonomic blockade is used to determine the intrinsic heart rate (see later).3
Exercise testing
Exercise testing to assess chronotropic incompetence is of value in patients with exertional symptoms (see later).
Electrophysiological Testing
Noninvasive testing is usually adequate in establishing the diagnosis of SND and guiding subsequent therapy. However, invasive EP testing can be of value in symptomatic patients in whom SND is suspected but cannot be documented in association with symptoms. In addition to assessing SND, EP testing can be useful in evaluating other potential causes for symptoms of syncope and palpitations (e.g., AV block, supraventricular tachycardia, and ventricular tachycardia).19,21
Electrocardiographic Features
Sinus Bradycardia
Sinus bradycardia (less than 60 beats/min) is considered abnormal when it is persistent, unexplained, and inappropriate for physiological circumstances. Sinus bradycardia slower than 40 beats/min (not associated with sleep or physical conditioning) is generally considered abnormal.3
Sinus Arrest
The terms sinus arrest and sinus pause are often used interchangeably; sinus arrest is a result of total cessation of impulse formation within the sinus node. The pause is not an exact multiple of the preceding P-P interval but is random in duration (Fig. 8-2). Although asymptomatic pauses of 2 to 3 seconds can be seen in up to 11% of normal individuals and in one third of trained athletes, pauses longer than 3 seconds are rare in normal individuals and may or may not be associated with symptoms, but they are usually caused by SND.3
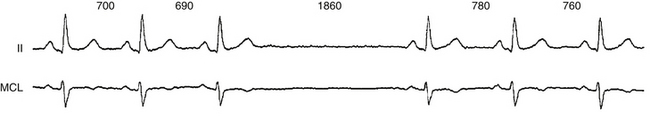
Sinoatrial Exit Block
Sinoatrial exit block results when a normally generated sinus impulse fails to conduct to the atria because of delay in conduction or block within the sinus node itself or perinodal tissue. Sinoatrial exit block produces a pause that is eventually terminated by a delayed sinus beat or an atrial or junctional escape beat.28 In theory, sinoatrial exit block can be distinguished from sinus arrest because the exit block pause is an exact multiple of the baseline P-P interval. However, sinus arrhythmia causing normal beat-to-beat variations in the sinus rate often makes the distinction impossible. Furthermore, establishing the diagnosis of sinoatrial exit block versus sinus arrest is often of academic interest only.3
Exit block is classified into three types, analogous to those of AV block: first-degree, second-degree, and third-degree exit block.28 First-degree sinoatrial exit block is caused by abnormal prolongation of the sinoatrial conduction time (SACT). It occurs every time a sinus impulse reaches the atrium, but it is conducted with a delay at a fixed interval. This type of sinoatrial exit block is concealed on the surface ECG and can be diagnosed only by direct sinus node recording or indirect measurement of SACT during an EP study. Second-degree sinoatrial exit block is marked by intermittent failure of the sinus impulse to exit the sinus node. Type I block is viewed as Wenckebach periodicity of the P wave on the surface ECG, and it manifests as progressive delay in conduction of the sinus-generated impulse through the sinus node to the atrium, finally resulting in a nonconducted sinus impulse and absence of a P wave on the surface ECG. Because the sinus discharge is a silent event on the surface ECG, this arrhythmia can be inferred only, because of a missing P wave and the signs of Wenckebach periodicity seen with this type of arrhythmia. The increment in delay in impulse conduction through the sinus node tissue is progressively less; thus, the P-P intervals become progressively shorter until a P wave fails to occur. The pauses associated with this type of sinoatrial exit block are less than twice the shortest sinus cycle. Type II block manifests as an abrupt absence of one or more P waves because of failure of the atrial impulse to exit the sinus node, without previous progressive prolongation of SACT (and without progressive shortening of the P-P intervals). Sometimes, two or more consecutive sinus impulses are blocked within the sinus node, thus creating considerably long pauses. The sinus pause should be an exact multiple of the immediately preceding P-P interval. However, normal variations in the sinus rate caused by sinus arrhythmia can obscure this measurement. Third-degree or complete sinoatrial exit block manifests as absence of P waves, with long pauses resulting in lower pacemaker escape rhythm. This type of block is impossible to distinguish from sinus arrest with certainty without invasive sinus node recordings.21,29,30
Tachycardia-Bradycardia Syndrome
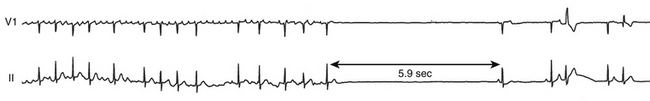
Tachycardia-bradycardia syndrome, frequently referred to as sick sinus syndrome, is a common manifestation of SND, and it refers to the presence of intermittent sinus or junctional bradycardia alternating with atrial tachyarrhythmias (Fig. 8-3). The atrial tachyarrhythmia is most commonly paroxysmal AF, but atrial tachycardia, atrial flutter, and occasionally AVN reentrant tachycardia or AV reentrant tachycardia can also occur.3
< div class='tao-gold-member'>

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


