Chapter 25 Ventricular Tachycardia in Nonischemic Dilated Cardiomyopathy
Pathophysiology
The diagnosis of nonischemic dilated CMP is established by the absence of significant (>75% stenosis) coronary artery disease and prior myocardial infarction (MI). Nonischemic dilated CMP is not a single disease entity. Valvular heart disease, hypertension, sarcoidosis, amyloidosis, Chagas disease, alcohol abuse, infections, and pregnancy, among others, need to be considered as possible etiologies. An underlying etiology for adult dilated CMP is found in only 50% of patients. The remaining 50% are considered idiopathic. Idiopathic dilated CMP is characterized by an increase in myocardial mass and a reduction in ventricular wall thickness. The heart assumes a globular shape, and there is a pronounced ventricular chamber dilation and atrial enlargement.1
To date, 33 genes have been linked to nonsyndromic familial dilated CMP. Notably, the frequencies of dilated CMP mutations in any one gene are low (<1% to 8%), and a genetic cause is identified in only 30% to 35% of familial dilated CMP cases. Although usually nonsyndromic, dilated CMP can be included in syndromic disease involving various organ systems, most commonly skeletal muscle disease (muscular dystrophy).2
As compared with sporadic cases of idiopathic dilated CMP, familial CMP patients are younger and tend to have higher left ventricular ejection fraction (LVEF) and more significant myocardial fibrosis. In patients with idiopathic dilated CMP, the proposed diagnostic criteria for the familial form of the disease are the existence of two or more affected family members, or of one first-degree relative with a documented history of unexplained sudden death before 35 years of age. In most cases, proof of a genetic cause of a CMP has a limited impact on the treatment of the index patient, but can have important implications in regard to family screening and genetic counseling. Dilated CMP in patients who do not have a known family history may also have a genetic basis.3
In contrast to ischemic heart disease, the electrophysiological (EP) substrate for sustained monomorphic ventricular tachycardia (VT) in patients with nonischemic dilated CMP is not clearly defined. Although bundle branch reentry (BBR) VT is identified as the VT mechanism in a significant percentage of patients with monomorphic VT in the setting of nonischemic CMP, the majority (80%) of VTs appear to originate from the myocardium and are due to scar-related reentry rather than BBR.4 BBR VT is discussed separately in Chapter 26.
Myocardial fibrosis, myocyte disarray, and membrane abnormalities are important factors in the substrate causing VT in patients with dilated CMP. Sustained VT is associated with more extensive myocardial fibrosis and nonuniform anisotropy involving both the endocardium and epicardium, compared with patients without sustained reentry. The reentry circuits are typically associated with regions of low-voltage electrograms, consistent with scar. Catheter mapping studies of patients with nonischemic CMP point to reentry around scar deep in the myocardium, near the ventricular base and in the perivalvular region, as the underlying mechanism for VT. Studies of explanted hearts with dilated nonischemic CMP have found inexcitable fibrosis creating regions of conduction block and surviving myocardium providing the substrate for potential reentry circuits. Slow conduction through muscle bundles separated by interstitial fibrosis can cause a zigzag path and promote reentry. Furthermore, patients with nonischemic CMP and predominance of scar distribution involving 26% to 75% of wall thickness (as quantified by magnetic resonance [MR] imaging) are more likely to have inducible VT. Delayed-enhancement MR imaging typically reveals nontransmural scar areas often distributed in the basal portion of the ventricular free wall or basal to midportion of the septum. Sustained VTs are observed more frequently in patients having a greater volume of hyperenhanced areas and greater number of hyperenhanced segments, and nontransmural scar tissue was present at the VT circuit exit site in the majority of patients.5,6
The scar and fibrosis resulting from nonischemic etiologies are distinctly different from post-MI scar; hence, the reentrant circuit may have different anatomical and functional properties that affect propagation. Compared with post-MI VT, the scar tends to be smaller and less confluent, the total number of the transmural scar segments is significantly smaller, and with less endocardial involvement in nonischemic CMP.4 Whereas ischemia produces a predictable wavefront of necrosis progressing from subendocardium to epicardium (and scar areas larger endocardially than epicardially), usually confined to a specific coronary vascular territory, scars in nonischemic CMP have been shown to have a predilection for the midmyocardium and epicardium. In contrast to the dense scar with isolated surviving myocardial bundles, scar in nonischemic CMP is patchy and may have fewer fixed boundaries and protected channels or isthmuses, which can alter the extent of local conduction slowing.7
Clinical Considerations
Epidemiology
Ventricular arrhythmias, both symptomatic and asymptomatic, are common in patients with nonischemic dilated CMP. Nonsustained VT can be observed in 30% to 50% of patients, but its incidence decreases significantly after optimization of medical treatment.8 However, syncope and SCD are infrequent initial manifestations of the disease. The incidence of SCD is highest among patients with indicators of more advanced cardiac disease who are also at highest risk of all-cause mortality. Although VT, ventricular fibrillation (VF), or both are considered the most common mechanism of SCD, bradycardia, pulmonary embolism, electromechanical dissociation, and other causes account for up to 50% of SCDs in patients with advanced heart failure.
Risk Stratification
LVEF has remained the most studied and the most powerful predictor and is the primary method currently used in clinical decisions for the prevention of SCD in patients with heart failure. Depressed LVEF is also a powerful predictor of cardiac mortality. On the basis of the results of large clinical trials, in clinical practice an LVEF of 35% or less has become the primary criterion used for prophylactic ICD placement. The use of LVEF as the predominant risk stratifier has serious limitations, however, because LVEF lacks sensitivity for prediction of SCD. Even a low LVEF (<20%) may not have high positive predictive value for SCD. Clinical factors such as functional class, history of heart failure, nonsustained VT, age, LV conduction abnormalities, inducible sustained VT, and atrial fibrillation influence arrhythmic death and total mortality risk and, consequently, potentially influence the prognostic value of a depressed LVEF. Therefore, patients with an LVEF greater than 30% and other risk factors may have a higher mortality and a higher risk of SCD than those with an LVEF less than 30% but no other risk factors.9
PVCs and nonsustained VT correlate with the severity of cardiac disease and occur in the majority of patients with severe LV dysfunction. This limits the usefulness of ventricular arrhythmias as risk stratifiers as they would be expected to be sensitive but not specific. Additionally, the presence and characteristics (frequency, length, and rate) of nonsustained VT do not appear to predict increased risk of subsequent life-threatening ventricular arrhythmias in patients with severe LV impairment receiving optimal medical treatment. Nevertheless, it has been suggested that the presence of nonsustained VT may be more specific in the individual with better LV systolic function. Nonsustained VT significantly increases the risk of malignant ventricular arrhythmias in the subgroup with an LVEF greater than 35%. In these patients, even without worsening LV systolic function and symptoms, survival free from malignant ventricular arrhythmias is similar to that of patients with an LVEF less than 35% with or without nonsustained VT.8
Cardiac MR can be used to evaluate the presence and magnitude of nontransmural scar tissue. Patients with nonischemic CMP and sustained VT typically have a greater volume and number of hyperenhanced (scar) areas compared with those without sustained VT.5
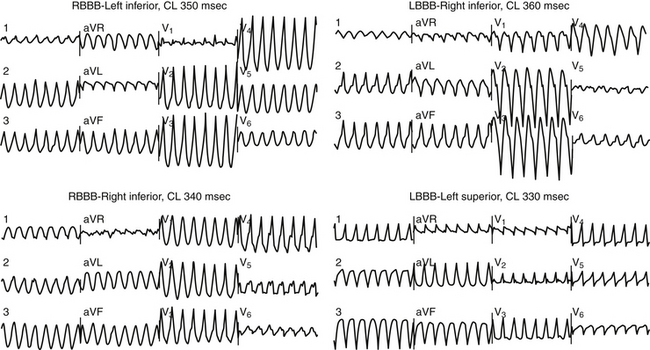
Patients typically have wide QRS complexes during the baseline rhythm, often with left bundle branch block or a nonspecific intraventricular conduction defect. Prolonged QRS duration has been associated with increased mortality in heart failure patients, but association with SCD has not been proven. Recently, fragmentation of the QRS complex on the 12-lead surface electrocardiogram (ECG) (filter range, 0.15 to 100 Hz; AC filter, 60 Hz, 25 mm/sec, 10 mm/mV) has been found to potentially predict increased risk of appropriate ICD therapies as well as a higher combined endpoint of ICD therapy and mortality in nonischemic CMP patients who received an ICD for primary and secondary prevention. The usefulness of this parameter needs further evaluation.10 During VT, QRS complexes are typically very wide and fragmented; most patients have multiple QRS morphologies of VT (Fig. 25-1).11
Prolongation of the QT interval, QT dispersion, and QT variability have had mixed predictive results with limited clinical applicability at present. Furthermore, studies evaluating the association of abnormal heart rate turbulence and reduced heart rate variability and SCD in heart failure patients have had conflicting results.9
Microvolt T-wave alternans has relatively modest (0.22) positive predictive value for SCD in patients with dilated CMP. Previous studies suggested a high negative predictive value for primary prevention of SCD, and T-wave alternans was hypothesized to be a useful tool to differentiate between patients who would benefit from ICD implantation and those who would not. However, results from recent studies failed to support this hypothesis and strongly suggested that a negative microvolt T-wave alternans result should not be used to withhold ICD therapy among patients who meet standard criteria.9
EP testing plays a minor role in risk stratification because of low VT inducibility, low reproducibility, and poor predictive value of induced VT. Although induction of VT by EP testing has been shown to predict SCD, unfortunately failure to induce VT misses most individuals destined to die suddenly.1
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


