Chapter 29 Ventricular Tachycardia in Arrhythmogenic Right Ventricular Cardiomyopathy-Dysplasia
Pathophysiology
Arrhythmogenic right ventricular cardiomyopathy-dysplasia (ARVD) is an inherited primary disease of the myocardium characterized by ventricular arrhythmias and structural abnormalities of the right ventricle (RV). The hallmark pathological findings are progressive myocyte loss and fibrofatty (fibrous and adipose) tissue replacement, with a predilection for the RV, but the left ventricle (LV) and septum also can be affected. Fibrofatty replacement of the myocardium produces “islands” of scar that can lead to reentrant VTs, and these patients have an increased risk of sudden cardiac death (SCD), mostly secondary to ventricular tachycardia (VT).1
Molecular Genetics
The autosomal dominant nonsyndromic ARVD-1 to ARVD-12 as well as the two rare recessive forms (Naxos disease and Carvajal syndrome) have been mapped to 12 genetic loci, with mutations in eight gene loci identified (Table 29-1). Although some identified gene mutations such as RYR2 may be phenocopies of ARVD, a common theme of desmosomal protein mutations has been emerging. In fact, five of the eight gene loci encode desmosomal proteins (desmoplakin [DSP], plakophilin-2 [PKP2], desmoglein-2 [DSG2], desmocollin-2 [DSC2], junctional plakoglobin [JUP]) and have been found in association with the ARVD phenotype in up to 50% of cases. Among the known genes, mutations in PKP2 appear to be the most common causes of ARVD, accounting for approximately 20% of cases. Mutations in DSG2 and DSP each account for approximately 10% to 15% of cases. Therefore, ARVD is currently considered, at least in a subset, a disease of the cardiac desmosome.2,3 The majority of these mutations are insertion/deletion or nonsense mutations, which are expected to cause premature termination of the encoded proteins.1
TABLE 29-1 Known Genetic Loci for Arrhythmogenic Right Ventricular Cardiomyopathy-Dysplasia
| LOCUS NAME | CAUSATIVE GENE | MODE OF INHERITANCE |
|---|---|---|
| ARVD-1 | Transforming growth factor-β3 | Autosomal dominant |
| ARVD-2 | Cardiac ryanodine receptor (RYR2) | Autosomal dominant |
| ARVD-3 | Unknown | Autosomal dominant |
| ARVD-4 | Unknown | Autosomal dominant |
| ARVD-5 | Transmembrane protein-43 | Autosomal dominant |
| ARVD-6 | Unknown | Autosomal dominant |
| ARVD-7 | Unknown | Autosomal dominant |
| ARVD-8 | Desmoplakin (DSP) | Autosomal dominant |
| ARVD-9 | Plakophilin-2 (PKP2) | Autosomal dominant |
| ARVD-10 | Desmoglein-2 | Autosomal dominant |
| ARVD-11 | Desmocollin-2 | Autosomal dominant |
| ARVD-12 | Plakoglobin (JUP) | Autosomal dominant |
| Naxos disease | Plakoglobin (JUP) | Autosomal recessive |
| Carvajal syndrome | Desmoplakin (DSP) | Autosomal recessive |
Mutations in genes encoding nondesmosomal proteins also can potentially cause ARVD. Alterations of the 5′ and 3′ regulatory elements of TGFB3, encoding transforming growth factor-β3, have each been reported. More recently, a mutation in TMEM43, encoding a transmembrane protein with ties to an adipogenic transcription factor, was reported as the cause for ARVD-5, a subtype of ARVD with prominent LV involvement. Mutations in RYR2, encoding the cardiac ryanodine receptor (the major calcium release channel of the sarcoplasmic reticulum in cardiomyocytes, also implicated in familial catecholaminergic polymorphic VT), result in a form of arrhythmogenic cardiomyopathy (ARVD-2) characterized by exercise-induced polymorphic VT that does not appear to have a reentrant mechanism, occurring in the absence of significant structural abnormalities. Patients do not develop characteristic features of ARVD on the 12-lead electrocardiogram (ECG) or signal-averaged ECG, and global RV function remains unaffected. ARVD-2 shows a closer resemblance to familial catecholaminergic polymorphic VT in both etiology and phenotype; its inclusion under the umbrella term of ARVD remains controversial.2–4
Pathogenesis
Cardiac desmosomes are specialized, multiprotein complexes in the intercalated disks that anchor intermediate filaments to the cytoplasmic membrane in adjacent cardiomyocytes, thereby forming a three-dimensional (3-D) scaffolding and providing mechanical strength and cohesion of cardiomyocytes in the beating heart (Fig. 29-1). Cardiac desmosomes are also responsible for regulating transcription of genes involved in adipogenesis and apoptosis as well as maintaining proper electrical conductivity through regulation of gap junctions and calcium homeostasis. Cardiac desmosomes are composed of three groups of proteins: the cadherin family, the Armadillo family, and the plakin family. The cadherin family is composed of three desmocollins and three desmogleins, which are primarily responsible for anchoring the structure to the membrane. The Armadillo family is composed of plakoglobin and three plakophilins, which form the core structure and possess signaling capabilities. The plakin family is composed of DSP, envoplakin, periplakin, plectin, and pinin, which are responsible for the attachment of the desmosomes to intermediary filaments.1,5
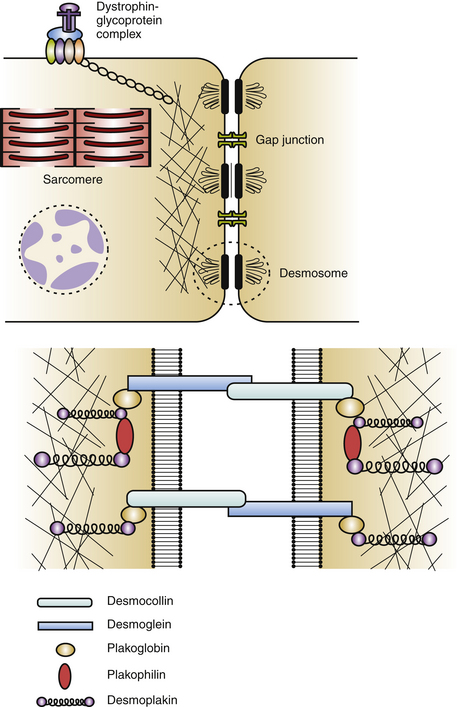
The mechanisms by which the affected desmosomes cause myocyte apoptosis, fibrogenesis, adipogenesis, and slow ventricular conduction, thus leading to impaired RV function and increased arrhythmogenicity, remain to be determined. Mutations in desmosomal genes alter the number or integrity of desmosomes and thereby lead to impaired mechanical coupling and failure of cell-to-cell adhesion structures during exposure to physical strain, resulting in cardiomyocyte detachment and degeneration, with subsequent inflammation and replacement by fibrofatty tissue. Fibrofatty replacement is a nonspecific repair process also observed in the muscular dystrophies. The architecture of thin surviving myocardial bundles within the fibrofatty tissue creates lengthened conduction pathways, conduction slowing at pivotal points, and conduction block. All factors contribute to activation delay and can create an electrophysiological (EP) substrate for reentry and VT. Ventricular dysfunction can ensue in the later stages owing to progressive myocyte detachment and death.6
Additionally, mutations in desmosomal proteins can impair expression of interacting proteins at the intercalated disk (e.g., gap junction or ion channel proteins), giving rise to impairment of intercellular conductance and promoting ventricular arrhythmogenesis, even in the absence of fibrofatty tissue replacement. Impaired desmosomal structure and function also can affect other cell-to-cell contact structures in the myocardium. In particular, connexin-43 remodeling in the gap junctions, which contributes to delayed conduction and ventricular arrhythmogenesis, is often observed in ARVD patients, and these potential electrical conduction abnormalities may favor arrhythmic events that characterize the disease and predispose patients to high risk of SCD.2–4
Pathology
Regardless of the mechanism, the patchy replacement of the RV myocardium by fibrofatty tissue provides a substrate for reentrant ventricular arrhythmias. The most striking morphological feature of the disease is the diffuse or segmental loss of RV myocytes, with replacement by fibrofatty tissue and thinning of the RV wall. Patchy inflammatory infiltrates can be present in areas of myocardial damage. Fibrofatty replacement usually begins in the subepicardium or midmural layers and progresses to the subendocardium. Only the endocardium and myocardium of the trabeculae may be spared. The sites of involvement can be localized and in early disease are often confined to the so-called “triangle of dysplasia”—namely, the RV outflow tract (RVOT), RV apex, and inferolateral wall near the tricuspid valve. RV aneurysms (Fig. 29-2), and segmental RV hypokinesia are typical. Diffuse myocardial involvement leads to global RV dilation. However, the fibrofatty pattern of ARVD is limited not only to the RV; the disease also can migrate to the LV free wall (commonly observed in advanced disease), with a predilection for the posteroseptal and posterolateral areas, with relative sparing of the septum.2–4
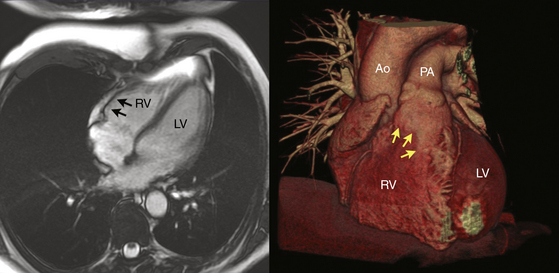
Despite the widespread impression that ARVD is universally a progressive disease process, a recent report found that the extent of the endocardial scar as measured by bipolar voltage mapping in patients with ARVD and VT remained relatively stable, despite progressive RV dilation.7
Clinical Considerations
Clinical Presentation
Four clinicopathological stages of ARVD can be considered: the early “concealed” phase, followed by the “overt electrical disorder,” the “phase of RV failure,” and finally, the phase of “biventricular failure.” Although ARVD is a genetically transmitted disease, it is associated with a long asymptomatic lead time and individuals in their teens may not have any characteristics of ARVD clinically or on screening tests. Early ARVD is often asymptomatic (the “concealed” phase), occasionally associated with minor ventricular arrhythmias and subtle structural changes. Nonetheless, these patients are still at risk of SCD, especially during intense physical exertion. With disease progression, the “overt electrical disorder” typically causes symptomatic ventricular arrhythmia and more obvious morphological abnormalities detectable by imaging. Further disease extension results in RV dilation and dysfunction (the “phase of RV failure”), precipitating symptoms and signs of right heart failure. Unless SCD occurs, progressive impairment of cardiac function can result in biventricular heart failure late in the evolution of ARVD, usually within 4 to 8 years after typical development of complete right bundle branch block (RBBB). End-stage disease is often indistinguishable from dilated cardiomyopathy and manifests with congestive heart failure, atrial fibrillation, and an increased incidence of thromboembolic events. Overall, judging the accurate position of the patient on the time scale of the spectrum can be difficult, and some patients may remain stable in the same phase of the disease for several decades.8
ARVD is one of the most arrhythmogenic human heart diseases known. Electrical instability is present at the very early stages of ARVD. Ventricular arrhythmias range from isolated ectopy to sustained VT or ventricular fibrillation (VF).8 Approximately 50% of patients with ARVD present with symptomatic ventricular arrhythmias, most commonly sustained and nonsustained VT, manifested by palpitations, dizziness, and/or syncope. The frequency of ventricular arrhythmias in ARVD varies with the severity of the disease, ranging from 23% in patients with mild disease to almost 100% in patients with severe disease. Ventricular arrhythmias characteristically occur during exercise; up to 50% to 60% of patients with ARVD show monomorphic VT during exercise testing.
Initial Evaluation
Electroanatomical voltage mapping, which seems to be an effective technique to detect RV low-voltage regions reflecting fibrofatty myocardial atrophy in patients with ARVD, has been shown to improve the diagnostic accuracy of myocardial biopsy by reducing the sampling errors. Endomyocardial biopsies are obtained from low-voltage areas, preferably from the border zone, in order to minimize the risk of perforation.9
Additionally, immunohistochemical analysis of conventional biopsy samples to detect a change in the distribution of desmosomal proteins can also improve the sensitivity and specificity of this diagnostic tool. Reduced immunoreactive signal levels of plakoglobin at intercalated disks were found to be a consistent feature in patients with ARVD but not seen in other forms of myocardial disease.10
Recognition of the problems in diagnosing ARVD and the fact that there is no “gold standard” or single test that is diagnostic of ARVD led to the formation of a task force in 1994 that proposed major and minor criteria to aid in the diagnosis, based on family history as well as structural, histological, functional, arrhythmic, and ECG abnormalities (Table 29-2).11 The diagnosis of ARVD is based on the presence of two major criteria, one major plus two minor criteria, or four minor criteria. It is important to recognize, however, that these criteria were defined retrospectively from aggregated series of referrals to tertiary care institutions dominated by the overt or severe end of the disease spectrum (i.e., symptomatic index cases and SCD victims) and have never been prospectively validated; therefore, their applicability is limited in situations where the prior probability of ARVD is different from the initial derivation set of patients. Consequently, the original task force criteria are highly specific but lacked sensitivity for early and familial disease. Furthermore, this diagnostic approach does not specify the preferred order of imaging techniques to examine and score RV morphology and function.5,11
TABLE 29-2 Original and Revised Task Force Diagnostic Criteria for Arrhythmogenic Right Ventricular Cardiomyopathy-Dysplasia









