Chapter 28 Ventricular Arrhythmias in Hypertrophic Cardiomyopathy
Pathophysiology
Changes in myocyte architecture lead to ventricular hypertrophy (Fig. 28-1). The degree and distribution of LV hypertrophy vary markedly. LV hypertrophy can be asymmetrical or symmetrical. The symmetrical form of hypertrophic CMP accounts for more than one-third of cases and is characterized by concentric thickening of the LV with a small ventricular cavity dimension. Asymmetrical septal hypertrophy is the most common variant, which is associated with thickening of the basal anterior septum, which bulges beneath the aortic valve and causes narrowing of the LV outflow region. However, isolated segmental hypertrophy may affect the LV apex (apical hypertrophic CMP) or any portion of the LV. The morphological pattern of LV hypertrophy is not closely predictive of the severity of symptoms or prognosis. Although LV apical aneurysms are observed only rarely (2%), they are associated with a higher rate of adverse disease consequences (10.5% per year) as compared with the general hypertrophic CMP population.1
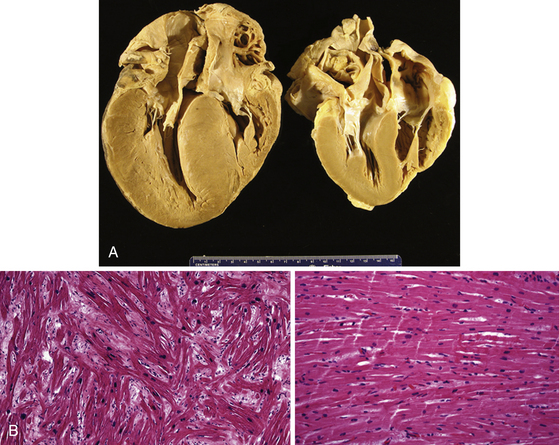
The arrhythmogenic substrate responsible for ventricular tachycardia (VT) occurrence in hypertrophic CMP has not been completely defined. Myofibrillar disarray as well as diffuse interstitial myocardial fibrosis or extensive scarring (likely caused by abnormalities of intramural coronary arteries and focal ischemia), which potentially predispose to disordered conduction patterns and increased dispersion of electrical depolarization and repolarization, have been suggested as factors contributing to ventricular arrhythmogenesis.2
Molecular Genetics
Hypertrophic CMP is familial in approximately half the cases and sporadic in the other half. The disease is transmitted as a Mendelian trait with an autosomal dominant pattern of inheritance and variable clinical penetrance. There is substantial diversity in the genetic causes of hypertrophic CMP. To date, nearly 900 different mutations have been reported in at least 24 genes encoding eight sarcomere proteins, including cardiac alpha- and beta-myosin heavy chains; cardiac troponins T, I, and C; cardiac myosin-binding protein C; alpha-tropomyosin; actin; titin; and essential and regulatory myosin light chains. Among these genes, mutations in MYH7, encoding beta-myosin heavy chain, and MYBPC3, encoding cardiac myosin binding protein-C, are the most common, each accounting for one-quarter to one-third of all cases.3,4 For each gene, several different mutations have been identified, and specific mutations are associated with different disease severity and prognosis.5
In addition, nonsarcomeric protein mutations in genes involved in cardiac metabolism (e.g., the gamma subunit of adenosine monophosphate [AMP]–activated protein kinase, PRKAG2, and lysosome-associated membrane protein 2 [LAMP-2], as in Danon disease), which are responsible for primary cardiac glycogen storage cardiomyopathies in older children and young adults, can be associated with a clinical presentation mimicking or indistinguishable from sarcomeric hypertrophic CMP. A high prevalence of conduction system dysfunction (with the requirement of permanent pacing in 30% of patients) characterizes PRKAG2 mutations. These diseases are often associated with ventricular preexcitation (Wolff-Parkinson-White syndrome). LAMP2 mutations are associated with early-onset LV hypertrophy (often in childhood) with rapid progression of heart failure and poor prognosis. These clinical entities are distinct from hypertrophic CMP caused by sarcomere protein mutations, despite the shared feature of LV hypertrophy. In addition, there can be genetic overlap between hypertrophic CMP and LV noncompaction.4
In infants and children, LV hypertrophy mimicking typical hypertrophic CMP caused by sarcomere protein mutations is often associated with congenital malformations and syndromes, inherited disorders of metabolism, and neuromuscular diseases, such as Fabry disease, Pompe disease, amyloidosis, carnitine deficiency, mitochondrial diseases, Friedreich ataxia, Noonan syndrome, and LEOPARD syndrome. Hypertrophic CMP can be distinguished from these disorders by dysmorphological examination, neuromuscular examination, metabolic screening, family information, and genetic testing.3,6
Clinical Considerations
Clinical Presentation
The clinical presentation of hypertrophic CMP is characterized by extreme variability in disease course, age of onset, severity of symptoms, and risk for SCD. Many patients are either asymptomatic or mildly symptomatic. The majority of patients present during adolescence or young adulthood, but symptoms can develop at any age. Symptomatic patients typically present with dyspnea, chest pain, palpitations, fatigue, orthostatic lightheadedness, presyncope, and syncope. Other complications include atrial and ventricular arrhythmias, infective endocarditis, and congestive heart failure. As noted, SCD can be the first manifestation of the disease.7
Shortness of breath, particularly exertional, is the most common symptom of hypertrophic CMP, occurring in up to 90% of patients, typically secondary to diastolic LV dysfunction. Syncope occurs in approximately 15% to 25% of patients, typically secondary to abnormal hemodynamic function (i.e., dynamic LV outflow obstruction) or, infrequently, secondary to cardiac arrhythmias.7,8 Atrial fibrillation (AF) is the most common arrhythmia observed in hypertrophic CMP, occurring in approximately 20% to 25% of patients, and is associated with an increased risk of stroke and thromboembolic complications.
Ventricular Arrhythmias
The predominant arrhythmia syndrome associated with hypertrophic CMP is sudden cardiac arrest, presumably due to polymorphic VT or VF. SCD occurs with an annual mortality rate of approximately 6% in referral-based populations and 1% in community-based studies. However, certain patient subgroups can have much higher rates, surpassing the American College of Cardiology/American Heart Association (ACC/AHA) guideline document definition of high risk for SCD (≥2% annual risk).9
Stable sustained monomorphic VT (SMVT) is rare, but can occur in patients with midventricular obstruction or apical LV aneurysms. The recurrence rate of VT is relatively high (56%) in hypertrophic CMP patients, and electrical storm can occur. During electrophysiological (EP) testing, induction of SMVT has a low reproducibility, and polymorphic VT and VF are often induced. The VT is commonly associated with a superior axis on the frontal plane, suggesting LV apical origins. An arrhythmic substrate, such as an aneurysm, can be important for the occurrence of stable SMVT.1,10
Appropriate implantable cardioverter-defibrillator (ICD) discharges in hypertrophic CMP patients occur annually in 10.6% of ICD recipients for secondary prevention after cardiac arrest (5-year cumulative probability, 39%), and in 3.6% of ICD recipients for primary prevention (5-year probability, 17%).11 Of note, arrhythmogenic events do not necessarily portend other adverse clinical outcomes. Specifically, appropriate ICD discharges do not appear to predict the occurrence of heart failure or the need for other invasive therapies (e.g., surgical myectomy, septal ablation).12
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


