Vasculitis
Bao Quynh N. Huynh
S. Louis Bridges Jr
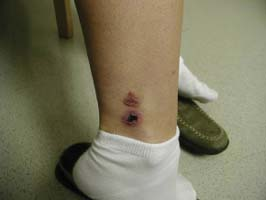 |
A 39-year-old female with an antinuclear antibody titer of 1:320, speckled pattern is referred to a rheumatologist for painful erythematous lesions on her lower extremities. Despite being on antibiotics for presumed cellulitis, her symptoms progressed, with additional similar lesions developing over the left ankle, left calf, right foot, and right lower leg. Her physical examination was remarkable for multiple 0.3- to 1-cm raised, palpable, tender purpuric lesions over the ankle and posterior aspect of the calves (Fig. 14.1). The remainder of the examination was within normal limits. Laboratory evaluation including complete blood count (CBC), chemistry profile, sedimentation rate, liver function tests, hepatitis serologies, and serum cryoglobulins was normal or negative. A skin biopsy of an active lesion was consistent with leukocytoclastic vasculitis. She was treated with prednisone, but had an incomplete response, so azathioprine was added. Over a course of several months, her lesions resolved completely, leaving only scarring (Fig. 14.1).
Clinical Presentation
Vasculitis is a heterogeneous group of disorders characterized by inflammation of blood vessels. One system of classification of vasculitides is based on the size of the predominant vessels involved (Table 14.1). For example, giant cell arteritis (GCA) involves large-sized blood vessels such as the aorta and its branches, whereas polyarteritis nodosa involves medium-sized vessels containing an internal elastic membrane, muscular media, and adventitia. Small-vessel vasculitis involves capillaries, and postcapillary venules and arterioles. Prior to the 1990s, a formal classification system of vasculitic syndromes did not exist because of a lack of consensus in evidence-based classification of individual patients with vasculitic syndromes (1). This was addressed by the American
College of Rheumatology (ACR) in 1990, which proposed criteria for the classification of seven different vasculitides (2). These criteria are not meant as diagnostic criteria, as they compared patients with different types of vasculitis, but not patients with other systemic or connective tissue diseases.
College of Rheumatology (ACR) in 1990, which proposed criteria for the classification of seven different vasculitides (2). These criteria are not meant as diagnostic criteria, as they compared patients with different types of vasculitis, but not patients with other systemic or connective tissue diseases.
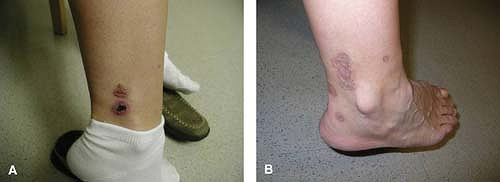 Figure 14.1 Skin lesions at presentation (A) and after resolution (B). By permission of Devore AE and Jorizzo JL. Chapter 39: Cutaneous small vessel vasculitis. In: Vasculitis, 2nd ed. Ball GV and Bridges SL Jr., eds. New York: Oxford University Press, Inc.; 2008. |
Table 14.1 Classification of More Common Forms of Vasculitis, Emphasizing the Predominant Size of Involved Vessels | ||
|---|---|---|
|
The ACR classification criteria do not include microscopic polyangiitis (MPA) or consider antineutrophil cytoplasmic antibodies (ANCA) as diagnostic criteria. In 1994, the Chapel Hill Consensus Conference (CHCC) produced definitions for vasculitis (3) and included MPA in its classification criteria. They also recognized that histological data would not be available for all patients, especially when the clinical condition of the patient might preclude obtaining appropriate biopsies. Furthermore, the sample might not be representative or the salient histological features may not be found because of sampling error. The classification scheme in Table 14.1 embodies features of both ACR and CHCC criteria, and is now widely used for epidemiological studies.
Epidemiology of Vasculitis
The vasculitides affect individuals of all ages, but are predominately seen in the extremes of age groups. Furthermore, the exact etiology is unknown, but it has been demonstrated to be multifactorial with factors such as ethnicity, genes, gender and ultraviolet light, infections, toxins, drugs, smoking, and surgery influencing disease expression (1).
Large-Vessel Vasculitis
Giant Cell Arteritis
There is an increasing incidence with age, with very few cases occurring in those younger than 50 years of age. There is a greater incidence in women, with a female-to-male ratio of around 2:1 (1). Giant cell arteritis is more common in Caucasians than in African-Americans and Hispanics. Interestingly, the incidence of GCA varies with latitude with increasing incidence with higher latitude.
Despite intensive investigation, no specific infectious agent has been identified. As the immune system ages it becomes more vulnerable to infections. Reinfection with the human parainfluenza virus (HPIV) was associated with the onset of GCA. Viruses such as hepatitis B, herpes zoster, Epstein–Barr virus (EBV), herpes simplex virus (HSV 1 and 2), respiratory syncytial virus, and adenovirus have been thought to cause GCA; however, there has been no evidence supporting the association between these organisms and the development of GCA (1).
Takayasu’s Arteritis
This form of vasculitis occurs worldwide, but it is more common in Asia. The annual incidence in most populations is 1 to 3 per million, with the peak age of the onset of disease in the third decade. The disease is more common in women (1).
Medium-Vessel Vasculitis
The annual incidence of polyarteritis nodosa (PAN) is 2 to 9 per million, with the highest incidence of 16 per million in Kuwait and 14.8 per million in Japan. Where there has been more attention to hepatitis B vaccination, the prevalence of PAN has decreased.
The prevalence of Behçet’s syndrome is highest in the regions extending from Eastern Asia to the Mediterranean basin and lowest in Western countries. The highest prevalence is seen in Turkey with 80 to 370 cases per 100,000 people. In Asian countries such as Japan, Korea, China, and Middle Eastern countries such as Iran and Saudi Arabia, the prevalence is between 13.5 and 20 cases per 100,000 people. Behçet’s syndrome is more common among females in Asian countries and among males in Middle Eastern countries. It occurs frequently in third and fourth decade of life.
Medium- to Small-Vessel Vasculitis
The incidence of antineutrophil cytoplasm antibody (ANCA)-associated (Wegener’s granulomatosis (WG), Churg–Strauss syndrome, and MPA) is approximately 10 to 20 per million per year, with onset peaking between ages 65 and 74 years (5). Wegener’s granulomatosis—a disease with necrotizing granulomata of respiratory tract, necrotizing vasculitis, and focal glomerulonephritis—is more common in men and is rarely seen in children (0.3 per million per year). Various studies have demonstrated that WG is not as common as microscopic angiitis in non-European populations, that is, Japanese and Chinese. Familial cases of WG have been reported, albeit rare. HLA DPB1*0401 has been associated with WG. Environmental factors such as seasonality and drugs have been previously thought to contribute to the development of WG. However, studies conducted by Lane et al. did not confirm the seasonal effect on developing this disease. No single drug has been shown to lead to the development of WG; however, certain drugs—including cocaine—can result in ANCA-associated conditions that mimic primary ANCA-associated vasculitis. Staphylococcus aureus infections have been associated with WG and a higher risk of relapse (5).
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


