Fig. 21.1
Schematic representation of gene targeting in mice. Embryonic stem cells (ES) are collected from mouse blastocysts (1). ES are cultivated in vitro (2) and a targeting vector is introduced by electroporation. The targeting vector contains fragments of DNA that are homologous to the endogenous gene (3). Homologous recombination occurs between the targeting vector and the endogenous gene (4). ES carrying the recombined gene are selected (5). The targeted ES are injected into blastocysts (6). The blastocysts are implanted into foster mothers (7) and they give birth to chimeric mice (8). The breeding between chimeric mice produces mice heterozygous for the targeted gene and wild-type mice (9)
If the mutated gene is a null allele, a global knockout for that targeted gene is then generated. Global knockouts provide direct insight into the physiological role of the ablated gene product. Moreover, novel actions of the targeted genes can emerge because, unlike transgenic models, global knockout models are not limited to a particular tissue or system. A disadvantage of the global knockout approach is that deletion of genes that are essential for early development may result in early lethality. On the other hand, because of functional redundancy, many knockout strains do not exhibit an obvious phenotype. In this case, creation of double or even triple knockouts may be necessary. Another consideration is that global knockouts usually contain a modified allele in which the selectable cassette used to screen the ES colonies is often retained in the locus of interest. In this case, this could have effects on neighboring genes.
If the mutated gene carries loss-of-function or gain-of-function mutations, a global knock-in for that targeted gene is then generated.
Instead of using a “targeted” strategy, global knockouts can be generated using a high-throughput mutagenesis strategy. One of the most widely used strategies involves the production of random insertional mutations in ES cells using vectors that contain a promoterless reporter gene.
21.2.2.2 Tissue-Specific Knockout Models
Conventional gene targeting generates a modified allele in all cells of the mouse from fertilization on; therefore, it is an extremely useful tool for investigating gene function during development and adulthood. However, if the inactivation of the target gene results in early embryonic lethality, the functions of the gene in specific tissues cannot be studied. Further, universal gene targeting can make it difficult to distinguish direct effects of ablating a gene in a particular tissue from the more indirect effects of ablating the gene in all tissues. The Cre recombinase-loxP (Cre-loxP) system was developed to overcome these limitations and to inactivate genes in a conditional manner in the living mouse as well (Orban et al. 1992; Sauer 1998). “Conditional” gene targeting refers to a gene modification in the mouse that is restricted either to certain cell types (tissue specific), to a specific developmental stage (temporally specific), or to both (Lewandoski 2001).
Table 21.1
Knockout and transgenic mouse models in the intervertebral disc
Gene of interest | Knockout | Annulus fibrosus | Nucleus pulposus | References |
|---|---|---|---|---|
GDF-5 | Universal | Abnormal | Abnormal | Li et al. (2004) |
Col2a1 | Universal | Abnormal | Normal | Sahlman et al. (2001) |
Col1a1 | Universal | Abnormal | Normal | Sarver and Elliott (2004) |
Col9a1 | Universal | Abnormal | Normal | Boyd et al. (2008) |
Allen et al. (2009) | ||||
Biglycan | Universal | Abnormal | Abnormal | Furukawa et al. (2009) |
Danforth’s short tail (Sd) | Universal | Abnormal | Absent | |
Sickle tail (Skt) | Universal | Abnormal | Abnormal | Semba et al. (2006) |
Sox5 and Sox6 | Universal | Deficiency of the inner annulus | Absent | Smits and Lefebvre (2003) |
Pax-1 | Universal | Abnormal | Abnormal | Wallin et al. (1994) |
Pax-9 | Universal | Abnormal | Abnormal | Peters et al. (1998) |
Has2 | Conditional | Abnormal | Abnormal | Roughley et al. (2011) |
Knockout in cartilage | ||||
c-Jun | Conditional | Normal | Abnormal | Behrens et al. (2003) |
Knockout in axial skeleton, sclerotome, notochord | ||||
Ext1 | Conditional | Abnormal | Abnormal | Mundy et al. (2011) |
Knockout in the developing joints | ||||
Smoothened | Conditional | Normal | Abnormal | Choi and Harfe (2011) |
Knockout in the notochord | ||||
Tgfbr2 | Conditional | Deficiency of the inner annulus | Normal | Jin et al. (2011) |
Knockout in growth plate chondrocytes and inner annulus fibrosus cells in the postnatal stage | ||||
Wnt/β-catenin | Conditional | Abnormal | Abnormality due to degeneration of the growth plate | Kondo et al. (2011) |
Knockout in axial skeleton |
The regional and temporal specificity provided by conditional gene targeting allows a better analysis of the gene function in different ways. The tissue specificity allows the study of a gene in a particular cell lineage without being influenced by gene loss in adjacent tissues, as the rest of the embryo is genetically wild type. Moreover, the temporal specificity does not permit the organism to adapt to the genetic change, as the wild-type gene product was previously present. Therefore, compensatory responses, which can alter interpretation of conventional germ line mutations, are mitigated, providing a more precise link between genotype and phenotype. Lastly, if null mutations lead to a severe or lethal phenotype during embryonic development, conditional gene targeting can also be used to investigate gene function at late embryonic stages or in adulthood.
To date, the Cre-loxP system is the best-characterized system for conditional gene inactivation in mice (Wilson and Kola 2001; Le and Sauer 2001). The Cre-loxP system is comprised of two elements: the Cre recombinase enzyme and a small stretch of DNA recognized by the recombinase (loxP site) (Stark et al. 1992; Van Duyne 2001). The Cre recombinase is produced by the bacteriophage P1 and is a member of the integrase superfamily of site-specific recombinases that cleave DNA at a distinct target sequence and ligate it to the cleaved DNA of a second identical site to generate a contiguous strand. The loxP site consists of 34 base pairs, a size that is unlikely to occur randomly in even the largest vertebrate genome and yet small enough to be effectively neutral toward gene expression when positioned in chromosomal DNA for genetic manipulations. The orientation of these target sites relative to each other on a segment of DNA directs the type of modification catalyzed by the recombinase; more specifically, in order to achieve excision of the intervening DNA, the two loxP sites must be oriented in the same direction.
Two separate mouse strains are typically generated and intercrossed for a conditional gene targeting experiment. One mouse strain expresses the Cre recombinase in selected tissues, depending on which promoter has been selected to drive recombinase expression; the mate carries a gene segment flanked (floxed) by the loxP sites (Fig. 21.2). The location of the loxP sites must be appropriately chosen so that the function of the gene is not affected and that deletion of the floxed gene segment will lead to inhibition of transcription and/or translation of the gene of interest or to the synthesis of a nonfunctional protein. In offspring, cells expressing the recombinase delete the target gene segment, while the target gene remains functional in cells of all other tissues where Cre is not expressed (Schipani 2002).
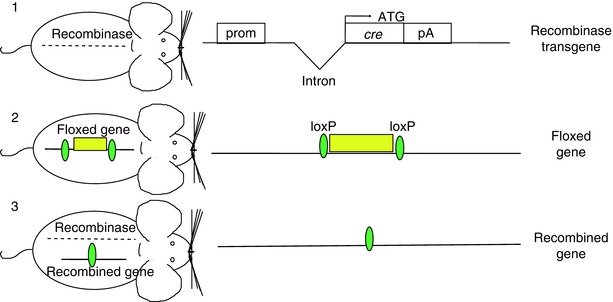
Fig. 21.2
Cre-loxP strategy. Two separate mouse strains are typically generated and intercrossed for a conditional gene targeting experiment. One mouse strain expresses the Cre recombinase in selected tissues (1); the mate carries a gene segment flanked (floxed) by the loxP sites (2). In offspring (3), cells expressing the recombinase delete the target gene segment
The ability to achieve site-specific recombination has revolutionized genetic analysis of skeletal cell function; it is important to bear in mind that it may be difficult to find a promoter that drives Cre expression with sufficient activity to result in complete excision of the target gene.
21.2.3 Studies of Disc Development and Function Using Genetically Modified Mice
21.2.3.1 Spontaneous Gene Mutations Causing Impaired Disc Function and/or Structure
Numerous animal models with spontaneous disc degeneration have been described. In these animals, the mutation is not artificially induced as the animals develop the pathological condition naturally.
The sand rat and the pintail mouse are the first spontaneous models that have been reported (Singh et al. 2005). As was discussed in Chap. 20, in the sand rat (Psammomys obesus), cysts and tears in the annulus fibrosus and even herniation of the nucleus pulposus are evident. These conditions resemble the pathological aspects of the degenerate human intervertebral discs. This phenotype may be related to aging but is mainly attributed to an altered metabolism. In fact, diabetic sand rats show a decrease in disc hydration, which leads to less advantageous biomechanical properties and eventually to degeneration of the disc (Silberberg et al. 1979; Ziv et al. 1992).
Another spontaneous disc degeneration mouse model is the pintail mouse (Anas acuta). The nuclei pulposi in the subcervical region have a low mucopolysaccharide content as in the degenerate human discs. This phenotype is stronger in homozygotes. This mouse model was the first evidence that deterioration of the human disc could indeed have a genetic correlate (Berry 1961).
Transcription factors of the Pax family play a major role in intervertebral disc development. Pax-1 is a transcription factor critically involved in various aspects of mammalian organogenesis (Chalepakis et al. 1992). It is expressed in the sclerotome and is implicated in the formation of ventral vertebral structures. Pax-1 is a critical modulator of the crosstalk between notochord and sclerotome in early development (Wallin et al. 1994). For further information on this gene, please see Chap. 3.
Mice carrying a naturally occurring mutation of the Pax-1 gene demonstrate impaired development of both the vertebral bodies and intervertebral discs (Wallin et al. 1994). In particular, both vertebral bodies and intervertebral discs may be either absent or severely deformed. The defects are especially evident in the lumbar region and in the tail. The malformations appear at an early embryonic stage and they affect both the sclerotome and the notochord. However, this phenotype most likely involves additional genes, as a targeted null mutation of Pax-1 causes a less severe phenotype (see below).
Watanabe et al. (1997) and, later, Wai et al. (1998) reported that the “cmd” (cartilage matrix deficiency) mouse carries an autosomal recessive mutation in the gene encoding aggrecan, which is expressed in both the nucleus pulposus and the annulus fibrosus. Homozygous mice for this mutation display dwarfism and cleft palate, and they die shortly after birth, whereas modest dwarfism, age-associated hyperlordosis, and disc herniation are typically found in heterozygous mice. Aggrecan is thus likely to be an extremely important molecule in the development and homeostasis of the intervertebral disc.
Li et al. (2004) analyzed instead a spontaneous loss-of-function mutation of growth differentiation factor-5 (GDF-5) in mice. This growth factor has been shown to play a role in a variety of musculoskeletal processes, including joint formation, endochondral ossification, and tendon and ligament maintenance and repair (Francis-West et al. 1999). Mutant mice carrying a spontaneous loss of function of GDF-5 are characterized by short limbs, abnormalities of the joints, and a reduction in the number of phalanges in the second through fifth digits (Merino et al. 1999). More importantly, these mice demonstrate abnormalities of both the annulus fibrosus and the nucleus pulposus, resembling changes seen in some animal models of disc degeneration. In particular, young adult mice displayed decreased water content of the nucleus pulposus as indicated by MRI analysis. Moreover, the mutant nucleus pulposus is smaller and the glycosaminoglycan content of the disc is diminished, although the amount of total collagen is not altered. In addition, the annulus fibrosus in these mutant mice is characterized by loss of the lamellar organization. All of these abnormalities can be corrected by treatment with recombinant GDF-5. Consistent with these data, conditional knockout (see subsequent paragraph) of GDF-5 further highlighted the importance of this growth factor in the biology of the nucleus pulposus.
21.2.3.2 Global Knockout of Genes Encoding Extracellular Matrix Proteins
Sahlman et al. (2001) reported that mice heterozygous for a Col2a1 null allele develop abnormalities in the vertebral bodies and in the disc. In particular, their vertebral end plates undergo premature ossification, which is associated with a mild grade of degeneration of the disc and with a significantly reduced ability to run, likely secondary to the discomfort caused by the anatomical abnormalities. Of note, collagen II is poorly expressed in the nucleus pulposus, though it is a classical marker of the fibrocartilage that forms the annulus fibrosus. Mice homozygous for a null mutation of Col2a1 show abnormalities in both vertebral bodies and intervertebral discs: the vertebral bodies enlarge gradually and they never initiate endochondral ossification; moreover, the notochord persists, leading to a failure in the development of the intervertebral disc (Aszódi et al. 1998).
Along these lines, Sarver and Elliott (2004) created a transgenic mouse with reduced collagen I expression (Mov13 strain). Collagen I is particularly abundant in the matrix of the outer annulus fibrosus. Mechanical testing provided evidence that the disc in Mov13 mutant mice is mechanically inferior to controls when subjected to compression and torsion tests. This finding suggests that collagen I in the intervertebral disc is particularly important in absorbing torsional loads.
Another essential matrix protein of the annulus fibrosus is collagen IX. This collagen acts as a bridge between the fibrillar collagens and the other components of the matrix; together with collagens II and XI, it forms a heterofibril, which stabilizes the cartilaginous tissue. Nakata et al. (1993) reported that mice in which a gene construct has been inserted to generate a truncated α1 chain of collagen IX develop chondrodysplasia, osteoarthritis, and corneal abnormalities. In particular, homozygous mice exhibit spine deformities characterized by shortening of the vertebrae, matrix disorganization within the annulus fibrosus, and end-plate irregularities (Kimura et al. 1996).
More recently, another study demonstrated that mice homozygous for a loss-of-function mutation of the Col9a1 gene have early-onset osteoarthritis around 6 months of age, secondary to degeneration both in the annulus fibrosus and in the end plate. Interestingly, in this mouse model, the changes in the intervertebral disc are already clearly detectable at 3 months of age and thus precede the degeneration of the vertebral end plate. Notably, disc degeneration occurs in the annulus fibrosus and it is mainly characterized by tears of this structure, whereas the nucleus pulposus is virtually unaffected (Boyd et al. 2008; Allen et al. 2009).
Furukawa et al. (2009) analyzed the function of another component of the extracellular matrix in the intervertebral disc by knocking out biglycan, which is a member of the family of small leucine repeat proteoglycans (SLRPs); SLRPs bind to TGF-βs, collagens, and other matrix proteins. In humans, biglycan is mostly expressed in the outer layer of the annulus fibrosus, while its concentration is very low in the nucleus pulposus. Advancing age leads to an increase in biglycan content in the early stages of degeneration and then, eventually, to a progressive decrease (Cs-Szabo et al. 2002). Previous studies showed that biglycan-deficient mice developed premature osteoarthritis (Ameye et al. 2002). In this study, the authors provide evidence that the absence of biglycan leads to an early degeneration of the intervertebral disc. Using morphometrical and histological analyses, the authors reported that the size of the nucleus pulposus decreases with age in the mutant mice and that, notably, at 6 months of age, mice display an abnormal proliferation of chondrocyte-like cells within the nucleus pulposus. Later, both the nucleus pulposus and the annulus fibrosus in biglycan-deficient mice are characterized by tears associated with “mucous degeneration” of the nucleus. Therefore, biglycan is likely to be an important factor in the maintenance of the intervertebral disc. There are different mechanisms to explain why loss of biglycan accelerates disc degeneration: its loss might cause instability of the extracellular matrix, or it might increase the mechanical stress on the intervertebral disc. The absence of biglycan may also inhibit TGF-β signaling, and this could suppress the damage repair response in the extracellular matrix.
21.2.3.3 Global Knockouts of Genes Affecting Disc Development
In recent years, numerous mouse mutants with abnormalities of the intervertebral disc have been generated by deleting genes of interest by homologous recombination. This knockout strategy has allowed researchers to define the role of different genes in disc development.
For instance, transcription factors important in chondrogenesis have also been shown to play a critical role in disc development and homeostasis. In particular, Smits and Lefebvre (2003) have provided evidence that Sox5 and Sox6 are expressed not only in the chondrocyte precursors that form the vertebral bodies and the annulus fibrosus but also in the notochord and are critically important for disc development. Sox5 and Sox6 encode two identical transcription factors (L-Sox5 and Sox6); moreover, Sox5 also encodes a short protein (Sox5) that lacks the N-terminal of L-Sox5. L-Sox5 and Sox6 are co-expressed in all cartilages and in a few other tissues. In vitro data have shown that these two transcription factors cooperate with Sox9 in the activation of the collagen II gene (Lefebvre et al. 1998). Moreover, Sox5 and Sox6 have redundant roles in chondrogenesis, and knockout mice for these two transcription factors are characterized by a severe chondrodysplasia (Smits et al. 2001). In a more recent work, Smits and Lefebvre analyzed the spine phenotype of mice lacking Sox5, Sox6, or both through RNA in situ hybridization, cell proliferation, and death assays. First, they showed that Sox5 −/− /Sox6 −/− mice lack the nucleus pulposus and that the annulus fibrosus has a deficient extracellular matrix. At earlier developmental ages, mutant embryos display an abnormal development of the notochord secondary to massive cell death. Analysis of the possible different genotypes led to the conclusion that Sox5 and Sox6 have redundant functions in notochord development, though Sox6 could be slightly more important.
Taken together, these findings demonstrate that Sox5 and Sox6 are not necessary in early notochord formation but in the survival of notochordal cells. Moreover, these data are consistent with the notion that the nucleus pulposus is derived from the notochord (Choi et al. 2008).
Homozygous Pax-1−/− mice display abnormalities in the shape of the vertebrae, and the intervertebral discs are replaced by a ventral cartilaginous rodlike structure (Wilm et al. 1998). In contrast to Pax-1 mutants, Pax-9−/− mutant mice do not exhibit morphological abnormalities of the axial skeleton (Peters et al. 1998). However, double mutants lacking both Pax-1 and Pax-9 have a more severe phenotype in both vertebral bodies and intervertebral discs when compared to single Pax-1 null mice (Peters et al. 1999). This study indicates that Pax-1 can completely compensate for the absence of Pax-9 during early development of vertebral bodies and intervertebral discs, whereas Pax-9 has a redundant role in the formation of these structures.
Related posts:
 Microenvironmental Control of Disc Cell Function: Influence of Hypoxia and Osmotic Pressure
Microenvironmental Control of Disc Cell Function: Influence of Hypoxia and Osmotic Pressure
 Gene Therapy Approaches for Disc Regeneration
Gene Therapy Approaches for Disc Regeneration
 Large Animal Models of Disc Degeneration
Large Animal Models of Disc Degeneration
 The Effects of Mechanical Forces on Nucleus Pulposus and Annulus Fibrosus Cells
The Effects of Mechanical Forces on Nucleus Pulposus and Annulus Fibrosus Cells
 Epidemiology of Lumbar Disc Degeneration
Epidemiology of Lumbar Disc Degeneration
 Proteoglycans of the Intervertebral Disc
Proteoglycans of the Intervertebral Disc
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


