CHAPTER 32 Tumors and Related Conditions
The discussion of musculoskeletal tumors is a broad and complex topic. Musculoskeletal tumors represent approximately 10% of all orthopaedic diagnoses and present a formidable clinical challenge because of their varying presentations and biological behavior. They usually come to clinical attention either because they are incidentally found or because they produce pain.
Most of the surgical reconstructive techniques (e.g., allografts, arthrodesis, and arthroplasty) presented have rather short follow-up to date, and therefore the interpretation of results should be kept in perspective. Many molecularly based biological findings relevant to musculoskeletal tumors have occurred since the late 1990s and are discussed briefly.1,2 Tumors involving the shoulder girdle are distinguished clinically by concern over a potentially increased risk of tumor recurrence and loss of function in this particular anatomic location as well as a definitely increased difficulty in achieving good functional recovery after proximal humeral resection.
HISTORICAL REVIEW
An exception to that rule was Samuel W. Gross (1837-1884), a well-known surgeon, pathologist, and anatomist at the Jefferson Medical College in Philadelphia, who authored one of the first works that attempted to deal with the classification of various sarcomas, their salient features, indications for treatment, and prognosis. Gross was one of the first persons in the world to identify sarcomas as a group of tumors distinctly different from carcinomas.3 With the discovery and development of radiographs (1893), various lesions of bone were beginning to attract attention. Gross was one of the first to appropriately identify sarcomas as locally invasive extremity tumors, with frequent metastases to the lungs and infrequently demonstrating lymphatic or hepatic metastases. These unusual lesions were associated with a history of trauma in half the cases and, according to Gross, required radical amputation or resection. In retrospect, his description of these first cases is remarkable for its clinical accuracy.
The scientific and technical developments in radiology, surgery, and medicine in the early 20th century resulted in significant advances in orthopaedics, which were reflected in improvements in the care of fractures, infections, and tumors. At that time, pathologists and surgeons such as John Ewing (New York), Ernest A. Codman (Boston), and James Bloodgood (Baltimore) became interested in various tumors of bone.4 Treatment of sarcomas varied greatly during those early years, but management of these unusual and difficult tumors gradually became more uniform as lesions were recognized histologically and radiographically as distinct entities. The same process of classifying sarcomas into histologic subtypes based on molecular subtype continues today.
Surgical treatment also improved with developments in pathology and radiology. Aggressive ablative surgery for sarcomas was first recommended by Gross in his classic article on sarcomas3 and was followed by various innovative surgical techniques.5,6 Syme of Edinburgh popularized scapulectomy for malignant tumors in 1856.7 Boris Linberg’s classic article in 1928 regarding interscapulothoracic resections8 for malignancies of the shoulder joint reported on aggressive surgery for skeletal tumors with limb salvage. Since these early reports, dramatic advances in imaging, chemotherapy, pathology, and surgical technique have resulted in improved survival and allowed more limb-sparing surgery.
In the 20th century, Dallas B. Phemister (1882-1951) of the University of Chicago was one of the first surgeons in North America to demonstrate a special interest in limb-sparing or “limb salvage” surgery as we know it today.9 Phemister reviewed the American College of Surgeons’ records for osteosarcoma in 1938 and found that only 4 of 86 extremity cases (4.6%) were treated with a limb-sparing resection.9 Other reports of limb-sparing surgery at that time described variable results in terms of morbidity and mortality.9,10 The popularity of limb-salvage surgery reached its zenith in the 1970s and 1980s with an emphasis on the need for appropriate tumor resection and good functional results.
The specialty of musculoskeletal oncology has crystallized from improvements in radiographic staging studies, chemotherapy, pathology, and surgery. One of the most significant developments has involved the evolution of a histologic grading system for sarcomas of bone and soft tissue that allows assessment of a patient’s prognosis according to the stage of the tumor and the proposed treatment.11–13 It is one of the only systems that appropriately reflects a patient’s prognosis based on the most significant determinants of that prognosis: the tumor’s mitotic index, histologic subtype, and histologic pleomorphism. It is a system that has demonstrated more predictability than previous grading systems have. New complementary DNA (cDNA) genotyping of tumors has allowed improved histologic subtyping and the ability to identify molecular defects.14
Sarcomas are unusual tumors that require complex treatment. Their rarity and heterogeneity have been major reasons for their haphazard treatment in the past. The difficulties associated with assigning specific subtypes to sarcomas has been well known for a long time. Even among expert pathologists the agreement rate for subtype is only 61% to 75%,15,16 and agreement varies by subtype. Recent advancements in description of the molecular phenotypes of these tumors and in assessment of their corresponding grade and molecular imaging17 have allowed more accuracy in tumor subtyping and in decisions regarding the treatment of high-grade tumors.
ANATOMY
Many anatomic considerations are involved in the treatment of musculoskeletal tumors. In the shoulder girdle, these considerations are amplified by the proximity of the brachial plexus and major vessels of the upper extremity to the humerus, scapula, and chest wall. Additionally, it has been suggested that the shoulder is more prone to intra-articular or pericapsular invasion by sarcomas than are other joints.18 This may be due to the relatively small joint size or thin synovial lining of the shoulder or because the biceps tendon provides a direct route into the joint. The implications of these anatomic points involve many aspects of the treatment and prognosis of shoulder neoplasms.
Although an evaluation of musculoskeletal tumors frequently refers to the various anatomic compartments of the region involved, the exact anatomy of the compartments about the shoulder remains poorly defined. The compartments of the shoulder (Fig. 32-1) include the deltoid compartment, the posterior scapular compartment (supraspinatus, infraspinatus, teres minor, and teres major), the subscapular compartment (subscapularis), the anterior pectoral compartment (pectoralis minor and major), the anterior humeral compartment (biceps and coracobrachialis), the lateral humeral compartment (brachialis), the posterior humeral compartment (medial, lateral, and long head of the triceps), and the intra-articular compartment of the glenohumeral joint. Little work has been done on the true containment or integrity of these compartments, and their boundaries are theoretical. Although they are anatomically based, their actual potential for containment remains untested.
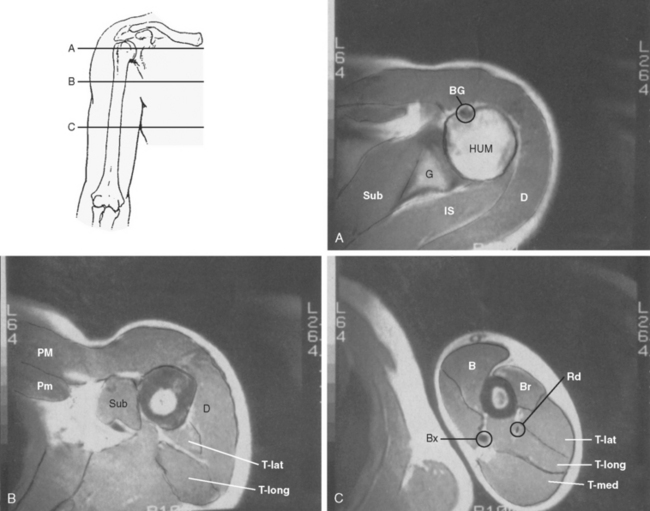
Another theoretical boundary for musculoskeletal tumors is the physis. It is thought the avascular environment of the physis as well as the presence of antiangiogenic factors inhibit tumor spread.19,20 Although the vast majority of lesions do not cross the physis, there are a few notable exceptions. These include aneurysmal bone cyst, chondroblastoma, osteogenic sarcoma, and osteomyelitis.
Many anatomic clues are helpful in making the initial diagnosis in patients with an unknown musculoskeletal lesion. For instance, Ewing’s sarcoma typically develops in the shaft or diaphysis of the humerus; it rarely occurs in the metaphysis of a long bone. On the other hand, the epicenter of an osteogenic sarcoma is rarely located in the shaft and is usually found in the metaphysis. Similarly, whether a lesion is intra-articular or extra-articular is important for several reasons. Intra-articular tumors are very unusual because most lesions have their epicenter in bone or in the soft tissues outside a joint. An intra-articular lesion is more likely to represent a degenerative, traumatic, or other non-neoplastic diagnosis. The fact that a tumor might involve a joint primarily or secondarily is significant from the point of view of treatment because it requires more-complex, extra-articular resection. Secondary involvement of a joint by an intraosseous malignancy is usually a late phenomenon associated with a longer diagnostic delay or a more-aggressive lesion and a worse prognosis (see the section “Staging and Classification of Tumors”).12,21
The shoulder girdle is unique in that it has one of the largest and best-defined muscle compartments in the body, that is, deltoid muscle. To function normally, the shoulder depends on a well-innervated deltoid and rotator cuff, in addition to adequate glenohumeral stability. The deltoid, like most muscle compartments, has anatomic subdivisions (acromial, clavicular, scapular), but grossly it is a well-defined muscle that is easily resectable, though extremely difficult to reconstruct functionally.
STAGING AND CLASSIFICATION OF TUMORS
The early classification system for musculoskeletal tumors was popularized by Lichtenstein, who classified tumors according to basic histologic categories.22 It was useful in identifying general trends in diagnosis and prognosis, but this descriptive histologic system had limited significance for determining adjuvant treatment (e.g., chemotherapy, radiation therapy) and prognosis. Today, most staging systems attempt to describe the anatomic extent of the disease as well as the presence or absence of metastases. They allow clinicians to compare clinical information in a clear and consistent manner. The stage of the tumor enables the physician to provide prognostic information to the patient.
Benign Tumors of Bone
One system that stages benign tumors relies on radiographic findings to assign a stage and uses an Arabic numeral system as opposed to the Roman numeral system used for staging malignant disease. Benign disease is denoted as stage 1, 2, or 3, depending on whether it is a latent, active, or aggressive tumor. A latent benign lesion does not show active growth and is confined to bone with minimal or no cortical involvement. A classic example is a nonossifying fibroma. An active lesion shows active growth but is confined within the compartment defined by the surrounding natural boundaries. There may be extensive cortical damage. Aneurysmal bone cysts and chondroblastomas are typically considered active lesions. An aggressive lesion has the potential to penetrate or violate natural boundaries, such as cortical bone or periosteum, and to remain locally aggressive without metastasizing. A giant cell tumor of bone is an ideal example. Campanacci has applied this staging system specifically to giant cell tumors in an effort to guide treatment.23
Theoretically, only malignant tumors (by definition) can metastasize; a contradiction in terms is presented by the ability of histologically “benign” giant cell tumors to “metastasize” to the lung in a few cases.24 Other aggressive benign tumors (chondroblastoma) have also demonstrated lung metastases in a small number of cases. Stage 2 and 3 lesions account for the vast majority of cases seen in clinical practice.
Malignant Soft Tissue and Bone Tumors
The most commonly cited staging systems for soft tissue sarcomas are the Memorial Sloan–Kettering Cancer Center (MSK) system, the American Joint Committee on Cancer Staging System (AJCC), and the system of Enneking and colleagues12 The MSK and AJCC systems take histologic grade, size, location, and presence of metastases into account, but they are organized differently.25,26 The highest category in both of these systems denotes the presence of metastases and therefore the worst prognosis. Critics of the AJCC system point out that the system has not been subjected to multi-institution scrutiny and is based on an inordinate number of nonextremity tumors.27
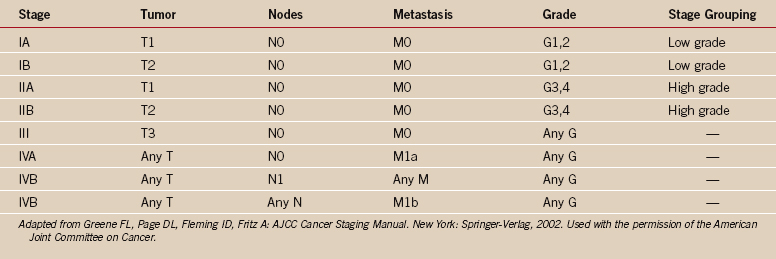
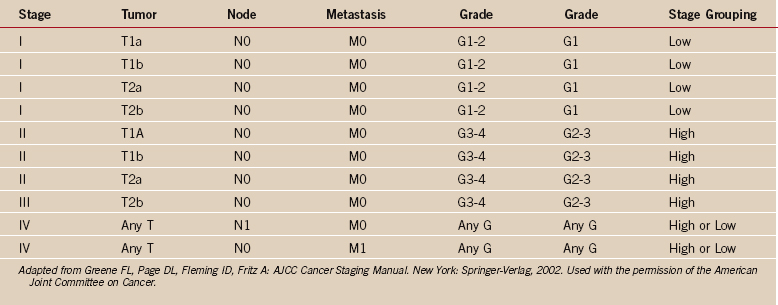
One of the greatest contributions to the improved treatment of sarcomas today has been the development of a staging system that assists in the selection of treatment, assessment of prognosis, and evaluation of results. Such a classification system was introduced by Enneking in 1980,12 adopted by the Musculoskeletal Tumor Society, and subsequently accepted, with modifications, by the National Institutes of Health Sarcoma Consensus Study Group as the staging system for all sarcomas.11 It represents a combined assessment of the histologic, or surgical, grade (G), the anatomic site of primary disease (T), and the presence or absence of metastases (M). Surgical grading is based on histologic assessment, with a lesion identified as benign (G0), low-grade malignant (G1), or high-grade malignant (G2) (Tables 32-1 and 32-2). The fundamental basics of the system can be applied to both soft tissue and bone sarcomas.
The original concept of the staging system as devised by Enneking represented a departure from the staging system of the American Joint Committee for Cancer Staging and End-Results,28 originally designed for evaluating various carcinomas. Enneking thought that the AJCC system correlated poorly with the natural history of sarcomas. He described the salient features of sarcomas with reference to their distinction from carcinomas and the significance of those features regarding staging.11,28,29
The extent of disease in the Enneking system is defined by the anatomic setting. Compartmentalization, or compartmental escape, is an important characteristic of sarcomas, in contrast to previous classification systems and other tumors (carcinomas). A tumor is considered intracompartmental if it is limited by natural boundaries such as fascia, bone, periosteum, or synovial tissue. A tumor is considered extracompartmental if it penetrates beyond its natural boundaries. It was postulated that the anatomic site (T) was the greatest factor in prognosis because it represented a composite of anatomic site, rate of growth, and delay in diagnosis. The primary extent of disease is limited by the natural boundaries of the anatomic compartment in which the lesion is located. A lesion located in the anterior of the thigh is contained by the fascial envelope of the quadriceps compartment until progressive growth causes it to extend beyond the natural barrier. When extension occurs, the patient has a worse prognosis and a higher risk of metastatic disease because of the presence of a more-aggressive tumor. Lesions that develop in poorly compartmentalized anatomic sites (e.g., groin, popliteal fossa, perivascular space) are, by the nature of that site, usually associated with a worse prognosis.
The histologic grading system proposed for sarcomas by the Enneking system was simplified to a two-grade system, that of high-grade versus low-grade histology. No allowance for intermediate-grade histology was made because there was no intermediate surgical treatment. This system required the pathologist to classify all sarcomas as either high-grade or low-grade lesions, which contradicts most classic sarcoma grading systems, in which high-, low-, and intermediate-grade histology is typically described. Grading remains a topic of controversy today, especially for soft tissue sarcomas, because they do occur as intermediate-grade lesions in certain cases.30 Intermediate-grade soft tissue tumors remain a treatment paradox regarding indications for chemotherapy. Tumor grading should not be based on the histologic type alone. The theory that some histologic diagnoses always represent high-grade lesions and a worse prognosis, regardless of their histologic grade, is not generally accepted.
The strength and weakness of the Enneking staging system is its simplicity. By emphasizing high-grade versus low-grade histology and by limiting the number of tumor stages (IA, IB, IIA, IIB, III), this system is simple enough to be used by a wide group of specialists and allows a variety of treatments. Malignant tumors are denoted as stage I, II, III, or IV (see Table 32-1),26 depending on the histologic grade (G), primary tumor extent (T), regional nodes (N), or the presence of metastases (M). Thus, a IA lesion is malignant, low grade, and intracompartmental. A IIA lesion is high grade and intracompartmental, and IIB is high grade and extracompartmental (see Table 32-1). Grade IV lesions are metastatic, regardless of the grade or anatomic site of the lesion. The system is somewhat different for soft tissue (see Table 32-2).31 Critics of the Enneking system cite the major weaknesses as being the overly simplistic nature of a two-grade system, the fact that subcutaneous sarcomas are difficult to define, and the lack of statistical validation of the system.27
The anatomic or surgical site classification (T) defines the primary lesion in relation to its position in the anatomic compartment of origin. Tumors are described as encapsulated (T0), intracompartmental (T1), or extra-compartmental (T2). This designation is based on the Enneking compartmental theory, in which an anatomic compartment is described as a space or potential space defined by natural boundaries.12 Tumors contained within an anatomic compartment can violate the boundaries of the compartment with growth—usually a sign of an aggressive benign or malignant tumor. Active benign tumors are typically well encapsulated (T0) and intracompartmental, whereas aggressive benign lesions may be poorly encapsulated but remain intracompartmental (T1). Low-grade malignant lesions are typically intracompartmental (T1), whereas extracompartmental lesions (T2) usually represent high-grade malignancies. Extracompartmental tumors can extend from one compartment into another or from one compartment into a surrounding extrafascial plane, or they can arise within a poorly compartmentalized, extracompartmental space. Poorly compartmentalized anatomic spaces include perivascular areas such as the subsartorial space of the common femoral artery, the popliteal fossa, the antecubital fossa, or the midhand, midfoot, axilla, or groin.
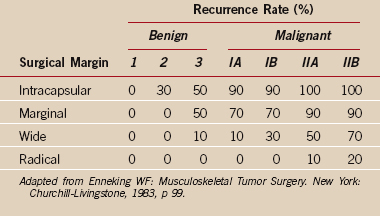
The stage of the lesion and the surgical margin achieved by a procedure are associated with a certain local recurrence rate as described in the work of Enneking (see the section “Surgical Margin” and see Table 32-5). These recurrence rates are based on an extensive retrospective review of the literature and reflect the risk of local recurrence after surgical resection without the use of adjuvant treatment. A benign aggressive (stage 3) lesion treated with a wide margin has a recurrence rate of 10% or less. This recurrence rate reflects surgical treatment alone and does not take into account the lower recurrence rate associated with surgery and adjuvant treatment, as performed for most malignant conditions. High-grade malignant tumors (IIB), such as a typical osteosarcoma, require at least a wide surgical margin that includes a surrounding cuff of normal tissue to prevent a local recurrence.
Thorough initial evaluation and staging, before treatment, remain the crucial ingredients for a successful outcome. Without these initial studies and an adequate and accurate biopsy, a successful treatment plan is unlikely. A universally accepted staging classification system is important to direct patient care and adequately assess clinical results regarding disease-free status. This initial staging philosophy remains one of the major contributions of the Enneking staging system to patient care. Unfortunately, widespread intraobserver and inter-observer disagreement continue to limit the clinical utility of musculoskeletal staging systems.
The staging evaluation involves an assessment by various radiographic studies to determine the precise anatomic extent of the primary disease, in addition to whether regional or distant metastases have occurred. Typical staging studies include plain radiographs, technetium bone scan, computed tomography (CT), MRI, and other studies that better define a lesion’s location. A total body bone scan is the best study to assess the extent of the primary bone lesion and the possibility of metastatic disease.32–34 CT scans are excellent for visualizing cortical geography and bone involvement at the primary site on a two-dimensional plane.12 CT scanning of the lung is routinely carried out in most institutions to assess possible pulmonary metastasis, and it is a more sensitive method than plain radiographs.
MRI is indicated for evaluating soft tissue disease, intramedullary bony disease, and spinal or pelvic lesions.35 The soft tissue or neurovascular margins are best assessed by MRI because it is much more sensitive than CT scanning for evaluating soft tissue margins. MRI does image the peripheral inflammatory reactive zone with a bright signal that might or might not contain a tumor. Similarly, the radiologist can over-read soft tissue margins when interpreting malignancies such as osteosarcoma and Ewing’s sarcoma35 because of the inability to distinguish inflammation from tumor on MRI.
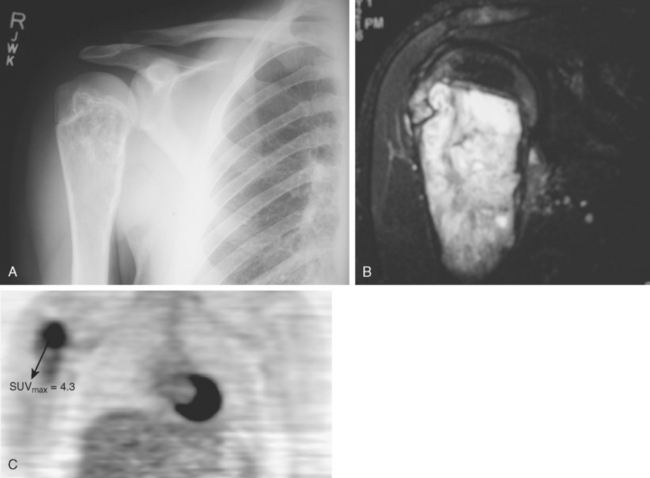
Tumor imaging has always been an essential ingredient for success in limb-salvage surgery, particularly for malignant tumors in which the malignancy involves bone or soft tissue. This importance of imaging is especially true for tumors in the shoulder area because of the increased complexity of proximal humeral and shoulder girdle tumors. Although MRI is clearly the optimal imaging modality for demonstrating anatomic detail and extent of tumor involvement in both bone and soft tissue malignancies, the current state of the art for assessing a tumor’s degree of malignancy and response to chemotherapy is quantitative positron emission tomography (PET) using fluorodeoxyglucose (FDG) (Fig. 32-2A to C). PET scans have the benefit of being quantifiable; that is, the technique can be validated to yield a specific numerical value, typically referred to as the standard uptake value (SUV). The SUV gives an indication of the tumor’s degree of malignancy and activity.36 This assessment is valuable in terms of grading tumors up front, evaluating response to treatment, assessing heterogeneity, and assessing local recurrences.37 Although PET scans are relatively early in their development for sarcoma applications, there is no doubt that these scans are valuable studies for assessing tumor grade, response to treatment, and the possibility of recurrence, and they are used routinely in most major centers today.
Molecular Biology
Despite these inherent challenges, some useful information has been gathered that can help us better understand tumor behavior. For example, overexpression of ErbB-2 or the multidrug resistance gene (MDR-1) has been associated with a poor prognosis for patients with osteosarcoma.38,39 A number of translocations are associated with certain types of sarcomas (Table 32-3). The characteristic t(11;22)(q24;q12) chromosomal translocation resulting in the EWS-FLI1 transcription factor in Ewing’s sarcoma has been shown to be a positive predictor of survival independent of tumor site, stage, and size.40 A gene expression signature has been identified in leiomyosarcomas that can predict the development of metastases.41 High levels of an apoptosis inhibitor gene, survivin, can portend a poor prognosis for osteosarcoma patients.42 Findings such as these might allow the clinician to tailor therapy based on the anticipated outcome of the disease.
TABLE 32-3 Chromosomal Translocations
| Tumor | Translocation |
|---|---|
| Ewing’s sarcoma | t(11;22)(q24;q12) |
| Synovial sarcoma | t(x;18)(p11;q11) |
| Extraskeletal myxoid | t(9;22)(q22;q12) |
| Chondrosarcoma | t(9;17)(q22;q11.2) |
| Clear cell sarcoma | t(12;22)(q13;q12) |
| Myxoid liposarcoma | t(12;16)(q13;p11) |
Classification of Tumors
Although the histologic classification of tumors has limitations in predicting the prognosis and directing treatment,17 it does serve a purpose in identifying tumor or sarcoma subtypes and their general tendencies. Knowledge of a tumor’s histologic type and the age of the patient are quite helpful in making a reliable tentative diagnosis in many patients, especially when the x-ray appearance is added to that information. The most common lesions of bone, cartilage, and soft tissue are described here in order to discuss their general histologic, radiographic, and clinical characteristics. Clinical features such as patient age, type of radiographic abnormality, and type of tissue involvement can, in many cases, lead to a significant and limited differential diagnosis.
Benign Osseous Lesions
Osteoid Osteoma
Benign osseous lesions of the shoulder are uncommon. Only 10% to 15% of cases of osteoid osteoma and osteoblastoma occur in the shoulder and, when they do occur, they favor the proximal end of the humerus or glenoid.43,44 Osteoid osteoma typically displays the classic symptom of night pain, which is relieved by salicylates. Radiographically, it is characterized by a large area of reactive bone surrounding a small, subcentimeter radiolucent nidus. On technetium bone scan, osteoid osteoma has impressive increased activity, and the central nidus can be visualized as a distinct cortical hole on CT scanning or tomography. Plain x-ray tomography can also be an effective diagnostic tool for localizing many osteoid osteomas. The differential diagnosis consists of osteoblastoma, osteomyelitis (Brodie’s abscess), intraosseous ganglion, stress fracture, and bone island. Histologically, this lucent nidus is a well-demarcated, small area of immature, very active osteoblastic tissue. Preoperative localization is an extremely important strategy to prevent intraoperative difficulty in locating these lesions and thus in minimizing local recurrences.
Treatment of an osteoid osteoma may be nonoperative or operative. Nonoperative treatment can only be considered if the diagnosis of osteoid osteoma is certain. NSAIDs can control symptoms; however, administration must be continued until spontaneous regression of the lesion occurs. This might not be acceptable to some patients. Other patients do not respond to medical therapy, experience side effects, or have a preexisting medical condition that contraindicates NSAID use. In these cases, curettage, with or without bone grafting, or en bloc excision are appropriate. CT-guided percutaneous radiofrequency ablation has been introduced as a successful, minimally invasive method of treating osteoid osteomas.45
Osteoblastoma
Some clinicians regard osteoblastoma as a larger version of osteoid osteoma (giant osteoid osteoma), and it is typified by a large lucent area (>2 cm) of osteoblastic tissue surrounded by a thin, sclerotic, reactive rim of bone.43 Radiographically, osteoblastoma is generally seen as a lucent lesion that has expanded the overlying cortex into a thin rim. The most common locations include the spine, femur, and tibia. As with osteoid osteoma, osteoblastoma may be difficult to localize radiographically and requires careful preoperative imaging to prevent recurrence. Osteoblastoma, unlike osteoid osteoma, also occurs in an aggressive (stage 3) form that is less well defined radiographically, has a high recurrence rate, and can have a histologic appearance that is difficult to distinguish from low-grade osteosarcoma. Technetium bone scanning and CT scans are good imaging techniques for examining both of these lesions.
Myositis Ossificans
Myositis ossificans is a benign, reactive, bone-forming process that develops intramuscularly or in the areolar tissues (tendon, ligament, capsule, fascia) adjacent to bone. It can occur with or without a history of trauma and, in the latter instance, may be referred to as pseudomalignant myositis ossificans of the soft parts.46 The pseudomalignant form is typically seen as a symptomatic enlarging soft tissue mass that develops in the second decade of life, and it occurs in the shoulder in 15% of cases.
In some patients, myositis ossificans may be confused with osteosarcoma or a soft tissue sarcoma, but these tumors do not demonstrate the same zonation or peripheral margination phenomenon, nor do they have the same radiographic characteristics. When the proper diagnosis is uncertain, optimal management includes a complete radiographic evaluation and careful clinical observation rather than a hasty or premature excision or biopsy (which can be difficult to interpret).47 The radiographic differential diagnosis for myositis ossificans includes extraosseous or parosteal osteosarcoma, synovial sarcoma, vascular lesions, and calcification of soft tissue secondary to necrosis, inflammation, or infection.
Malignant Osseous Lesions
Osteosarcoma
Classic osteosarcoma is a high-grade, aggressive tumor that develops in metaphyseal bone, typically as a stage IIB lesion and usually with an extraosseous soft tissue component present at initial evaluation.22,43,48 The typical patient has intrinsic bone pain at night that is typically unrelated to activity (Box 32-1). The average duration of symptoms at initial assessment is 3 to 6 months, which reflects the subtle nature of the preliminary symptoms and the need for early recognition of intraosseous pain and night pain as warning symptoms.49,50
BOX 32-1 Signs and Symptoms of Sarcoma
From Enneking WF, Spanier SS, Goodman MA: A system for the surgical staging of musculoskeletal sarcoma. Clin Orthop Relat Res (153):105-120, 1980.
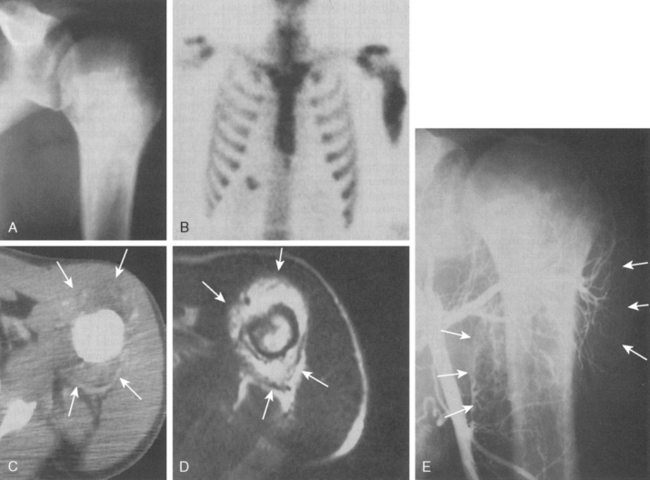
Approximately 10% to 15% of all osteosarcomas occur in the proximal part of the humerus, whereas 1% to 2% develop in the scapula or clavicle.22,48,51,52 The typical radiograph for osteosarcoma has a sunburst or osteoblastic pattern, with penetration of the adjacent cortex (Fig. 32-3A). Differential diagnosis includes aneurysmal bone cyst, Ewing’s sarcoma, osteoblastoma, giant cell tumor, and metastatic disease. Osteosarcomas usually have increased activity on bone scan (see Fig. 32-3B), and a hemorrhagic soft tissue mass is seen on CT scan (see Fig. 32-3C) and MRI (see Fig. 32-3D). Arteriography is no longer the technique of choice for evaluating soft tissue involvement but it may be performed to evaluate for major vessel involvement or for response to intra-arterial chemotherapy (see Fig. 32-3E).53 Variants of osteosarcoma other than the classic type include telangiectatic (vascular) osteosarcoma,54 secondary osteosarcoma (Paget’s disease or radiation induced), and various low-grade lesions such as periosteal and parosteal osteosarcoma.52,55,56 The basic histologic criterion for the diagnosis of classic osteosarcoma includes a malignant stroma-producing (spindle cells) tumor or immature neoplastic osteoid.22,48,51 The overall survival rate for patients with osteosarcoma at 5 years of follow-up is approximately 70% with appropriate chemotherapy and surgery.57 Prognostic factors for survival remain debatable; however, histologic necrosis at resection and the size of the tumor are probably the most significant variables.58
Benign Cartilaginous Lesions
Osteochondroma
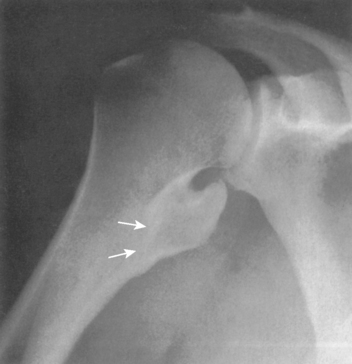
The incidence of cartilaginous tumors in the shoulder is second only to those occurring about the pelvis.59 Solitary osteochondroma, or exostosis, is the most common benign tumor of the shoulder; approximately a fourth of all exostoses occur in the proximal part of the humerus. Osteochondromas actually represent a developmental abnormality arising from the peripheral growth plate and are typically active, benign (stage 2) lesions during skeletal growth. The plain radiograph is usually diagnostic in demonstrating a smooth excrescence of metaphyseal cancellous bone that is confluent and continuous with normal metaphyseal bone (Fig. 32-4).
Exostoses can appear as pedunculated, stalk-like lesions or as flat, sessile lesions. Concern regarding a possible secondary chondrosarcoma can arise in adult patients with pain, an enlarging soft tissue mass, or intraosseous bony erosions. Evidence of a thickened cartilaginous cap (1 cm) on CT scan, in association with a soft tissue mass, pain, or radiographic evidence of possible malignant degeneration, suggests a secondary chondrosarcoma. Dedifferentiation is rare and associated with a poor prognosis.60 The risk of a secondary chondrosarcoma arising out of an exostosis is approximately 1% per lesion, although rates as high as 10% to 30% have been referred to in the literature regarding secondary malignancy in patients with multiple hereditary exostoses.61
Chondroblastoma
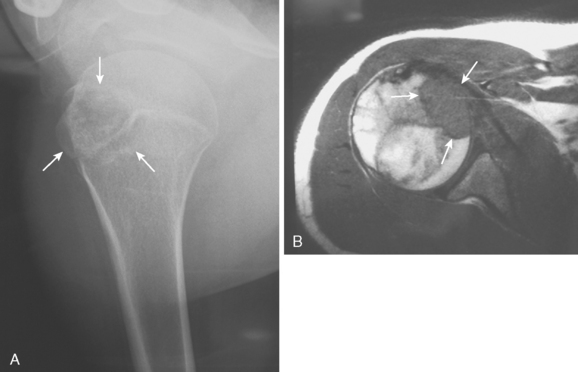
Chondroblastoma, or Codman’s tumor, is an unusual benign cartilaginous tumor that occurs in the proximal humeral epiphysis (25% of cases) as a round or oval lesion containing fine calcifications surrounded by a reactive bony margin.62–65 It occurs in the skeletally immature, and histologically it consists of aneurysmal tissue, chicken-wire calcifications, and immature paving-stone chondroblasts. Chondroblastoma occurs as an active, benign stage 2 lesion, although it also has a more aggressive stage 3 form. In very rare cases, metastatic disease to the lungs has been demonstrated.
Treatment usually involves extensive intralesional curettage, which results in a large subchondral defect of the humeral head that requires bone graft to prevent subchondral and cartilaginous collapse. An adjuvant agent such as hydrogen peroxide or cryotherapy can help to decrease local recurrence rates.66 The radiographic appearance of this epiphyseal lesion is usually typical (Fig. 32-5A and B), and it develops in adolescents or young adults. The differential diagnosis includes a simple cyst, eosinophilic granuloma, osteomyelitis, enchondroma, or an aneurysmal bone cyst.
Periosteal Chondroma
Periosteal chondroma is another benign cartilaginous lesion of the proximal end of the humerus that is usually located just proximal to the deltoid insertion of the lateral humeral shaft. It is typically manifested as a minimally symptomatic or asymptomatic mass that is radiographically evident as a sessile lesion with a distinct, well-defined margin of reactive cortex underlying the radiolucent cartilaginous mass.67 Marginal excision results in a cortical defect of the humerus that might or might not require bone grafting. Curettage is associated with a high recurrence rate. The differential diagnosis includes periosteal osteosarcoma, which does not have the well-defined underlying sclerotic cortex. Periosteal osteosarcoma is a more aggressive intracortical lesion that can extend into the medullary canal in a small percentage of cases.
Enchondroma
Enchondroma is a benign, central cartilaginous lesion that is most commonly found in the small tubular bones of the hand but also occurs in the proximal end of the humerus in 10% to 15% of cases.68–70 Enchondromas are usually solitary, but multiple, typically unilateral, lesions with extensive deformity can be seen in Ollier’s disease (enchondromatosis) (Fig. 32-6) or Maffucci’s syndrome (enchondromatosis with multiple hemangiomas of skin or viscera, or both).
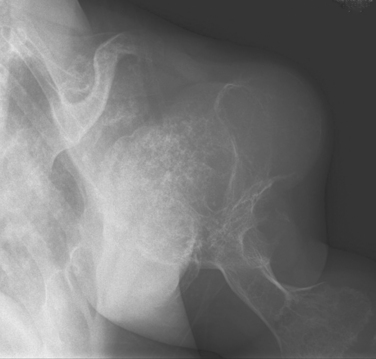
Treatment of an enchondroma is based on a thorough assessment of the presence of pain and risk of fracture. Lesions that are painless can be observed with serial radiographs. If a patient develops pain, or there is evidence of radiographic progression, then reevaluation is mandatory. The risk of malignant transformation of solitary lesions is extremely small. By contrast, the risk of malignant transformation is 25% to 30% in Ollier’s disease and even higher in Maffucci’s syndrome. Malignant degeneration is usually heralded by increased or new pain or by a sharp increase in tumor size.
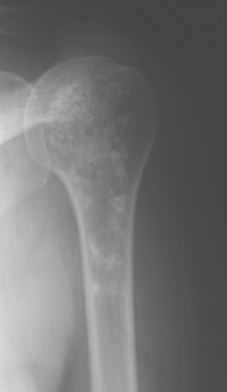
When an enchondroma occurs adjacent to a joint that is symptomatic for degenerative reasons, clinical assessment of bone pain related to the enchondroma may be difficult. This scenario is not uncommon, and it makes the initial evaluation of intraosseous cartilage tumors difficult because intrinsic bone pain is an important symptom suggesting a low-grade malignancy. Thus, the ability to distinguish intraosseous from intra-articular symptoms is a difficult but necessary challenge. The typical radiographic appearance of a benign enchondroma is that of a central lucent lesion with a well-defined bony margin and intrinsic calcifications. Figure 32-7 presents such a lesion in a 45-year-old woman with rotator cuff symptoms and a calcified benign cartilage lesion.
Malignant Cartilaginous Lesions
Chondrosarcoma
Chondrosarcoma can develop de novo as a primary chondrosarcoma, or it can arise out of a preexisting benign cartilage lesion and is then referred to as secondary chondrosarcoma. Secondary chondrosarcoma occurs in young adults, accounts for approximately 25% of all chondrosarcomas, and may be found in patients with a preexisting enchondroma, osteochondroma, multiple enchondromatosis (Ollier’s disease),64,71,72 or multiple hereditary exostosis.73 The radiographic evidence of a secondary, or low-grade, chondrosarcoma arising out of such a lesion includes enlarging radiolucent areas within the lesion or endosteal cortical erosions along the cortical margins (Fig. 32-8A and B). Technetium bone scans are typically moderately hot for both enchondroma and low-grade chondrosarcoma and are not helpful in distinguishing one from the other.
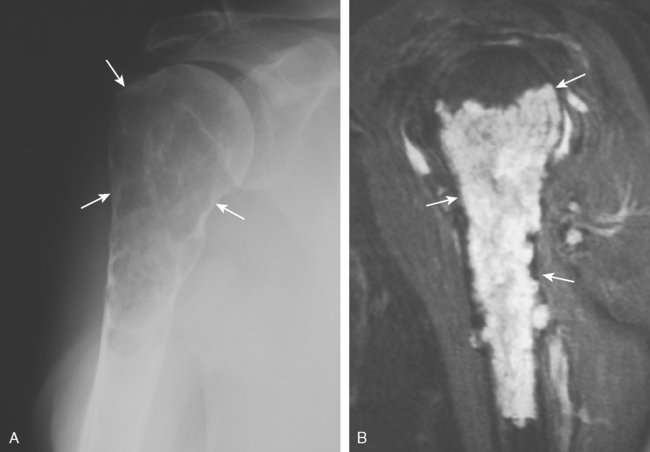
Microscopic evaluation of cartilage lesions is not diagnostic in a large percentage of cases. Histologic characteristics suggesting malignancy include cellularity, pleomorphism, and evidence of mitotic activity, such as double-nucleated lacunae. These findings are subtle, and the histologic evidence for low-grade chondrosarcoma versus enchondroma is often incomplete and incon-clusive.49,71 This confusion has led to use of the term grade one-half chondrosarcoma to describe cartilage tumors that are histologically borderline between low-grade chondrosarcoma and benign enchondroma. In most patients, the diagnosis of low-grade chondro-sarcoma is best made radiographically by assessing whether there is evidence of endosteal erosions created by active cartilaginous tumor growth. Low-grade, or secondary, chondrosarcomas are unique among intraosseous tumors in the difficulty in interpreting their light microscopic picture.
Primary chondrosarcoma is more commonly seen in the middle decades of life, and its incidence in the shoulder is second to that in the pelvis or hip joint.74–78 It is the most common primary bone malignancy to arise in the coracoid process.79 These tumors are typically manifested as intraosseous lesions with poorly defined margins and faint intrinsic calcifications. The differential diagnosis includes enchondroma, osteosarcoma, and metastases. Less commonly, a primary chondrosarcoma arises from the surface of a bone or joint. Its clinical and radiographic appearance is very subtle, and a diagnostic delay of 6 to 12 months is not uncommon.
Approximately two thirds of primary chondrosarcomas are also low grade and can have the appearance of benign, encapsulated cartilage lesions. High-grade lesions are more invasive, have a higher metastatic rate, and usually occur in long-standing lesions as a dedifferentiated chondrosarcoma.47,80–82 As a general rule, low-grade chondrosarcomas may be treated surgically with curettage and grafting, whereas high-grade tumors deserve surgical resection and reconstruction.
Synovial Dysplasias
Cartilaginous loose bodies typically arise out of a proliferative synovium in a reactive metaplastic process known as synovial chondromatosis (or osteochondromatosis).83 It most commonly affects large joints (knee, elbow, shoulder, hip) in young adults and results in multiple small, cartilaginous, intra-articular loose bodies as the process matures. In the few cases in which the nodules form a compact mass of cartilage, it may be confused with a low-grade, periarticular or juxta-articular chondrosarcoma. An intra-articular location favors the benign diagnosis of synovial chondromatosis; consequently, determining whether it is intra-articular or extra-articular is sometimes one of the preoperative goals. In such cases, MRI or CT scanning with or without arthrography might pinpoint the exact site of involvement. Synovial chondromatosis is typically a slowly progressive, degenerative disease that ultimately leads to joint destruction. It requires aggressive total synovectomy to prevent persistence or recurrence, and in older patients with degenerative disease it is well treated with joint excision and replacement. A few reports in the literature associate malignant transformation with long-standing synovial chondromatosis.84,85
Another disease associated with proliferating synovium is pigmented villonodular synovitis.86 It is generally associated with a boggy, inflammatory synovitis, with or without bony erosions, in adolescents or young adults. Histologically, it is an aggressive synovial-histiocytic process that defies description as inflammatory or neoplastic. Treatment requires aggressive complete synovectomy for the diffuse form of the disease. Various forms of radiation therapy have been used in some centers with acceptable early clinical results.87 Occasionally, joint degeneration is severe and arthroplasty is recommended.88
Miscellaneous Intraosseous Tumors
Simple Bone Cyst
Simple bone cysts, or unicameral bone cysts, occur most commonly in children between the ages of 4 and 12 years. Figure 32-9A demonstrates a simple cyst with a healing pathologic fracture in the humerus of an 8-year-old. The patient remained symptomatic after fracture healing and underwent percutaneous aspiration and injection of autogenous bone marrow and demineralized bone matrix. The cyst completely resolved (see Fig. 32-9B) and the patient returned to full activities 3 months later. Simple bone cysts are well-defined, central, radiolucent lesions arising in the metaphysis adjacent to the physis (active) and, with maturation, migrate distally into the diaphysis (latent). Occasionally, a fragment of the cyst wall breaks off and floats into the fluid-filled cyst cavity. Radiographically, this appearance is known as the fallen-leaf sign. Simple bone cysts typically involve the proximal part of the humerus (50%), contain straw-colored fluid, and may be confused with an aneurysmal bone cyst or, less often, fibrous dysplasia.10,31,89–92
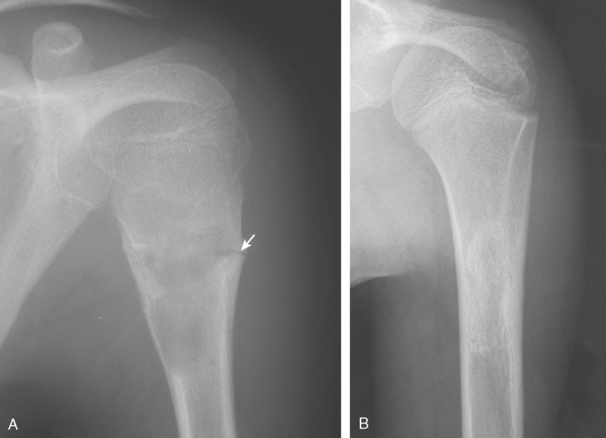
The treatments of choice for symptomatic lesions consist of pressure measurement of the cyst, aspiration, and either intraosseous steroid injection or injection of demineralized bone matrix with autogenous bone marrow.92–94 The result is complete healing of the cystic area in approximately 50% of cases and partial healing in 45%.94 Complete repair after injection is most common in more inactive cysts with lower pressure. Varying results in more-recent reports have cast some doubt on the efficacy of steroid injections for simple bone cysts, especially when associated with a venogram at the time of injection of dye into the lesion.31 Comparison of injection of steroids versus autologous bone marrow has yielded similar success rates; however, multiple steroid injections are often necessary to achieve healing.91,95
Recurrence or persistence of the cyst after surgical curettage and bone grafting occurs in approximately 30% of cases. This relatively high local recurrence rate may be lowered by the addition of liquid nitrogen freezing to the curettage.96 Some diagnostic overlap occurs between aneurysmal and simple cysts in children because some simple cysts can have hemorrhagic fluid and yet do not contain aneurysmal tissue. In general, if a cyst fractures, it is advisable to allow sufficient time for fracture healing before initiating treatment. This can abrogate the need for internal fixation.
Aneurysmal Bone Cyst
Aneurysmal bone cyst, nonossifying fibroma, and fibrous dysplasia are all benign lesions that can occur in the shoulder. The molecular biology of aneurysmal bone cysts has been elucidated and appears to involve up-regulation of oncognenes.97 Aneurysmal bone cysts are not uncommon in the proximal end of the humerus, but because of their widespread occurrence as a secondary lesion engrafted on other tumors (simple cyst, giant cell tumor, chondroblastoma, osteoblastoma), the true incidence is unknown. The radiographic hallmark is that of a lucent, expansile metaphyseal lesion. MRI shows the presence of fluid-fluid levels (Fig. 32-10). Treatment includes curettage plus bone grafting,98–100 which is associated with a recurrence rate of 20% to 30%. Open growth plates and young age are associated with a higher recurrence risk.101 Aneurysmal bone cysts can have an aggressive appearance, and a careful biopsy should be performed before curettage to exclude the possibility of telangiectatic osteosarcoma. It may be extremely difficult to distinguish these two entities on frozen section alone. In such cases, definitive treatment should be delayed until a final diagnosis is rendered.
Fibrous Dysplasia
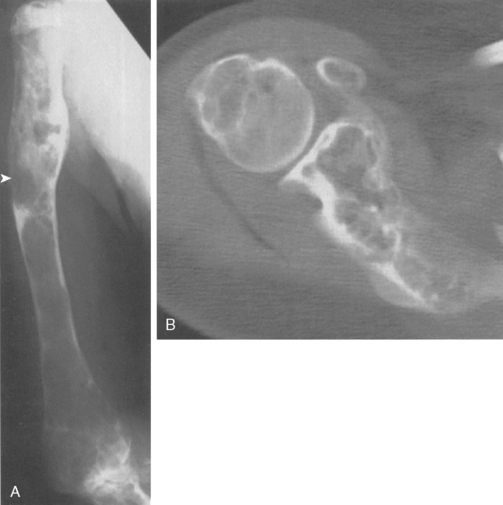
Fibrous dysplasia is a congenital dysplasia of bone that often surfaces as a painful lesion secondary to pathologic fracture, microfracture, or the subtle, intrinsic, diaphyseal weakness resulting from pathologic bone. The typical plain radiograph demonstrates a ground-glass density with cortical thickening. Figure 32-11A and B is the plain radiograph and CT scan, respectively, of the humerus of a 20-year-old woman with severe polyostotic fibrous dysplasia. She had a history of chronic pseudarthroses (see Fig. 32-11A) that had persisted despite bracing. When associated with symptoms or pathologic fracture, diaphyseal involvement usually requires intramedullary fixation rather than bone grafting because cancellous bone graft is consistently consumed by the dysplastic process and is ineffective in resolving the weakened dysplastic bone. There may be a role for medical therapy, bisphosphonates, in alleviating pain; however, randomized clinical studies are lacking.102 Histologically, fibrous dysplasia demonstrates a furnace of dysplastic bone activity with similar, impressive increased activity on bone scan.103–107 Immature islands of bone are seen in a fibrous stroma without osteoblastic rimming.
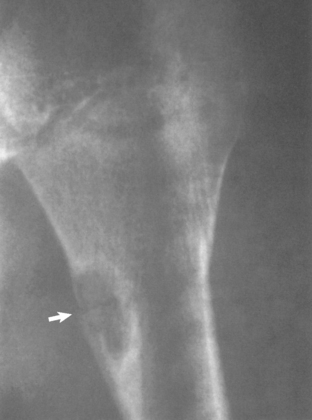
Nonossifying Fibroma
Nonossifying fibroma is a benign fibrous lesion that appears radiographically as an eccentric, well-defined, lucent lesion that has a scalloped border abutting the adjacent cortex (Fig. 32-12). It is more commonly found in the lower than the upper extremity. When the lesion is smaller than 4 cm, it may be referred to as a fibrous cortical defect. When longer than 5 cm or occupying more than half the transverse diameter of the bone, these lesions are at risk for pathologic fracture. The majority of nonossifying fibromas heal spontaneously and require no treatment. Treatment is reserved for lesions with atypical radiographs (requiring biopsy) or for symptomatic or larger lesions that require treatment to prevent a pathologic fracture.108
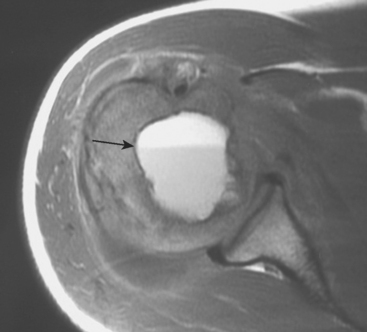
Giant Cell Tumor
Giant cell tumor (GCT) of bone is a benign, locally aggressive lesion. Although classically considered a benign tumor, GCT of bone has the potential for metastatic pulmonary spread in less than 3% of cases.109–116 Giant cell tumor of bone is a common lesion in people between 20 and 40 years of age that develops primarily in the distal end of the femur or proximal part of the tibia (60%-70%). It can also occur in the proximal end of the humerus in 5% to 10% of cases.
GCT is a radiolucent, epiphyseal–metaphyseal tumor that most commonly has a distinct bony margin and is often associated with extensive subchondral bone erosion.117 There can be bony expansion, cortical destruction, or frank extension of the tumor mass into the soft tissues. Periosteal reaction is uncommon unless there has been a prior pathologic fracture. Although most cases involve solitary lesions, the rare diagnosis of multifocal GCT of bone is usually associated with hand lesions and a slightly younger population. The radiographic differential diagnosis in an adult includes aneurysmal bone cyst, brown tumor of hyperparathyroidism, metastatic adenocarcinoma, lymphoma, chondrosarcoma, intraosseous ganglion, and osteomyelitis.
GCT is typically a Campanacci stage 2 active lesion (60% of cases) but also shows up as a more-aggressive stage 3 tumor in 20% of cases. Treatment alternatives for GCT include curettage, with or without local adjuvant treatment, or marginal resection. The local recurrence rate after curettage alone is 20% to 30% for active lesions versus 5% after marginal resection.23,109,118–120
Reticuloendothelial Tumors
Tumors of reticuloendothelial origin include a category of intraosseous lesions that arise from marrow stem cells and lesions of similar histology. They are also referred to as round cell or small, blue cell tumors. This category of tumors or abnormalities includes diagnoses such as leukemia, lymphoma,121 neuroblastoma, histiocytosis, rhabdomyosarcoma, Ewing’s sarcoma, infection, and, in adults, multiple myeloma and metastatic adenocarcinoma.
Multiple Myeloma
Multiple myeloma is the most common primary malignancy of bone and typically occurs in the middle decades of life; the shoulder girdle is involved in 5% to 10% of cases.110,122 The most common site of involvement is the axial skeleton, but multiple distinct lesions develop in the extremities in a significant number of patients and can require surgical stabilization to prevent impending fracture if medical treatment has failed. In patients who have a solitary intraosseous myeloma or plasmacytoma of the shoulder at initial evaluation, biopsy is indicated for diagnostic reasons.
Elevated serum calcium levels, anemia, serum protein electrophoresis, or a distinctly cold bone scan can suggest the diagnosis of myeloma before biopsy in a patient with a solitary lesion or unknown diagnosis. The overall prognosis is poor; however, newer treatments involving aggressive chemotherapy and plasma cell antibodies offer hope for the future. The development of new classes of bisphosphonates has had a positive effect on decreasing skeletal events and bone pain.123
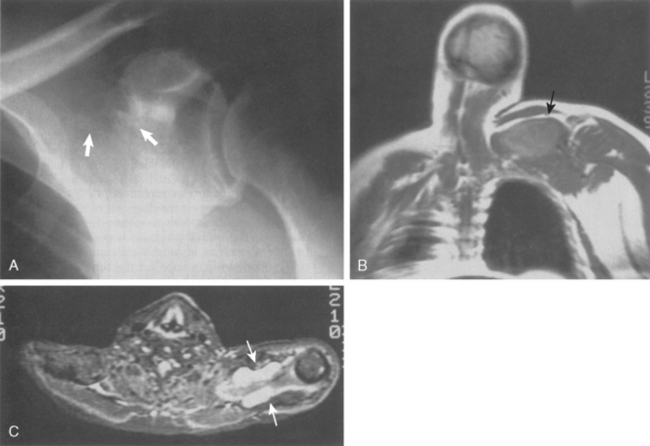
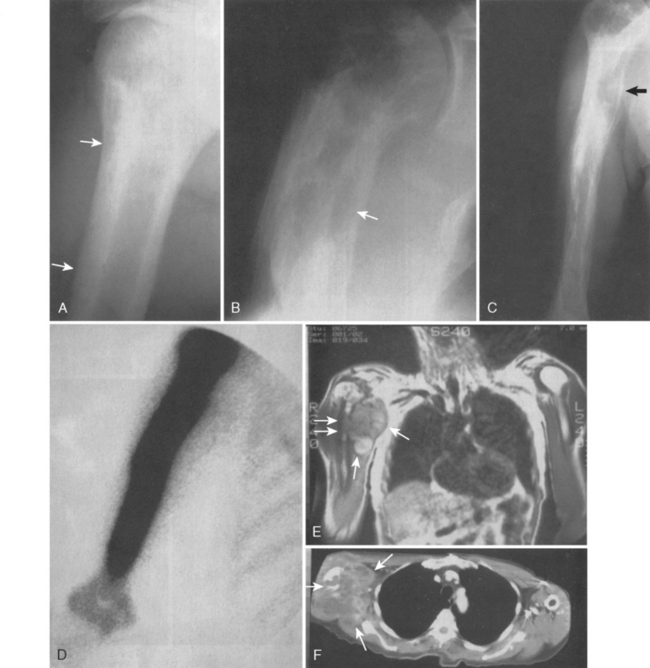
Figure 32-13A is the plain radiograph of a 42-year-old man in apparent good health but experiencing shoulder pain. Coronal (see Fig. 32-13B) and transverse (see Fig. 32-13C) MRI views demonstrate a suprascapular soft tissue lesion that extends anteriorly and posteriorly to the scapula. The preoperative diagnosis was a probable soft tissue sarcoma. Open biopsy was diagnostic for multiple myeloma with extensive bone disease. The patient died suddenly 1 week after the biopsy, with an undocumented serum calcium level. All patients with the diagnosis of myeloma need to have careful evaluation of their serum electrolytes for the possibility of hypercalcemia, which is usually heralded by altered mental status, fatigue, weakness, or nausea.
Ewing’s Sarcoma
The second most common intraosseous malignancy in adolescence is Ewing’s sarcoma, an aggressive marrow cell tumor that appears as a permeative diaphyseal tumor that is poorly marginated and typically associated with a large, soft tissue mass.29,124 Occasionally, the tumor permeates through the bone marrow and funnels through the canal. Differential diagnosis consists of osteomyelitis, osteosarcoma, lymphoma, eosinophilic granuloma, other round cell tumors, and neuroblastoma, especially in children younger than 2 years.
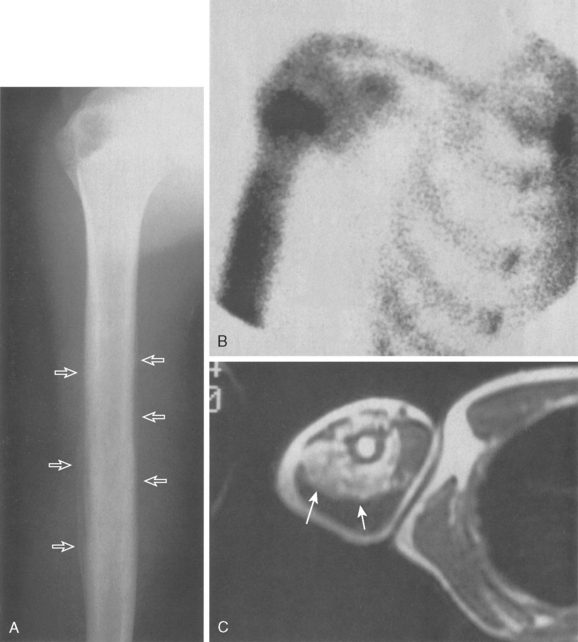
Figure 32-14A demonstrates such a permeative lesion in the humeral diaphysis of a 16-year-old with a typically hot bone scan (see Fig. 32-14B) and an associated soft tissue mass (see Fig. 32-14C). Although plain radiography is useful for diagnostic purposes, it is not reliable for determining the extent of intramedullary disease. For this reason, an MRI is mandatory before surgery to accurately determine the resection margins. Ewing’s sarcoma today is primarily treated with aggressive chemotherapy and surgical resection or radiation therapy, depending on the size and location of the primary lesion.
Miscellaneous Dysplasias
Gaucher’s Disease
Gaucher’s disease is an uncommon metabolic disorder of the reticuloendothelial system and glucocerebroside-glycolipid metabolism that affects the liver, spleen, and bone marrow.125 The disease has an increased incidence in the Jewish population and presents most commonly in the first 3 decades of life and without sexual preference. Patients typically present with cytopenia, hepatosplenomegaly, and bone pain.126 Bone pain is secondary to vascular thrombosis and most commonly occurs in the femoral head, with a high degree of bilaterality.
The disease in many ways represents a form of avascular necrosis of the bone. The humeral head is the second most common site of involvement, and radiographic changes include osteopenia, diaphyseal or medullary expansion, and cortical erosions. The differential diagnosis includes osteomyelitis in the acute setting and round cell tumors in the nonacute setting. Surgical treatment involves internal fixation for fracture prophylaxis or treatment of deformity, joint replacement in adults when indicated, and appropriate management of femoral head necrosis in children. Enzyme replacement is the mainstay of systemic therapy and has been shown to greatly decrease skeletal morbidity.127
Paget’s Disease
Paget’s disease (osteoporosis circumscripta, osteitis deformans) occurs after the fourth decade and has a slight preponderance in men.128 It is the second most common metabolic bone disorder in people older than 50 years. Geographically, it appears to have a higher incidence in Great Britain, Western Europe, Australia, and the United States, whereas it is relatively rare in India, Asia, and Africa. Paget’s disease develops most commonly in the pelvis, skull, lumbosacral spine, femur, and humerus. It can occur in a polyostotic or a monostotic form and is usually evident at the time of initial evaluation. The typical radiographic picture shows cortical thickening and rarefaction, followed by pathologic microfracture and diaphyseal bowing. The differential diagnosis in an adult includes metastatic adenocarcinoma, osteosarcoma, and osteomyelitis. Patients should be assessed by evaluation of serum alkaline phosphatase and urinary hydroxyproline levels, a total body bone scan, and a CT scan or MRI.
Most patients with Paget’s disease do not require surgical management. The minority of patients who do require surgery usually have musculoskeletal complaints related to deformity, facture, or altered joint mechanics. Patients with Paget’s disease undergoing orthopaedic surgery should, in general, be pretreated to decrease bleeding associated with the hypervascularity of the bone. Paget’s disease itself is best managed medically with bisphosphonates or calcitonin.129 Zoledronic acid is the most recent bisphosphonate (FDA approved in 2007) to be used for treatment of this condition.
Sarcoma arising out of Paget’s disease is characterized by a history of progressive pain and a bony lytic lesion with a soft tissue mass (Fig. 32-15A and B). Radiographically, Paget’s sarcoma of the scapulohumeral area is characterized by predominantly lytic changes in the humerus and purely sclerotic changes in the scapula.130 Pagetoid sarcoma is a rare variant of osteosarcoma with a very poor prognosis regardless of site or stage at presentation.131–133 Paget’s sarcoma is best managed by ensuring a radical surgical margin because of the diffuse nature of the process of Paget’s disease and the difficulty of assessing the extent of sarcomatous changes.
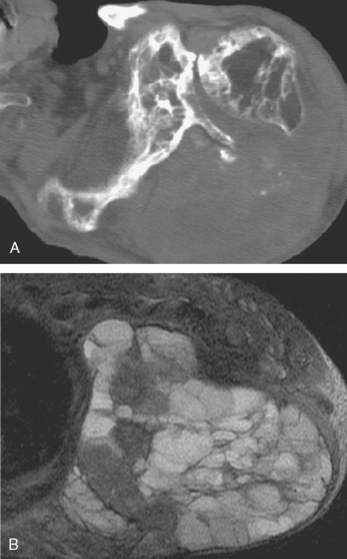
Figure 32-16A and B presents early and late radiographs of Paget’s disease in the proximal end of the humerus. The lytic lesion, combined with a history of increasing arm pain, served notice of an early secondary osteosarcoma that showed up 3 months later with a more impressive lytic lesion in the proximal part of the humerus (see Fig. 32-16C). Paget’s disease affected the full humerus, and the bone scan (see Fig. 32-16D) was of little help in demarcating bony margins or osseous involvement by this secondary, or pagetoid, osteosarcoma.22 MRI and CT scans again demonstrate the soft tissue and bony extent of disease in the proximal part of the humerus (see Fig. 32-16E and F).
Benign Soft Tissue Tumors
Ganglion
A ganglion is a common soft tissue tumor that is often confused with other cystic lesions. Ganglia have a thin lining but no true synovial capsule. They are filled with a characteristic gelatinous material. In the shoulder, they are often associated with degenerative conditions or a labral tear. MRI is the study of choice for evaluating these lesions and for determining the precise anatomic location. Typically, the MRI shows a rounded or lobular fluid signal mass with low signal on T1 images and high signal on T2 images. When located in the spinoglenoid notch, a ganglion may produce suprascapular nerve palsy secondary to nerve entrapment.134
Lipoma
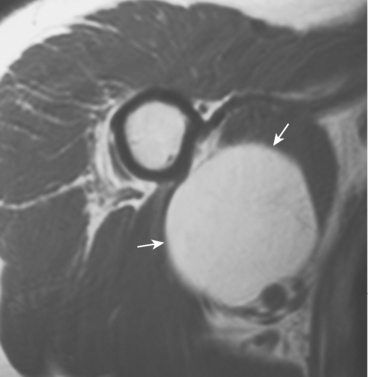
Lipomas can occur intramuscularly or within normal fat planes of the axilla or the subscapular or other perivascular spaces. They often appear in the anterior deltoid as a large, soft, nontender, intramuscular mass.135 A few lipomas are tender or firm or have an equivocal history of a change in size. On MRI or CT scan, a benign lipoma usually has a uniform, fatty consistency (Fig. 32-17). Clinically, a liposarcoma has a firmer, denser consistency than a lipoma does. If a lipoma feels very dense or firm clinically, MRI should be performed for further evaluation. If MRI demonstrates areas of distinctly different density, a biopsy should precede marginal excision to exclude the possibility of a liposarcoma.
Hemangioma
Hemangiomas typically appear as enlarging intramuscular lesions in a child or young adult. They are best visualized by MRI and typically have a serpiginous configuration of vessels. If they are intimately involved with a major vessel, they should also be evaluated with an arteriogram. These lesions do not usually pose diagnostic or surgical problems, with the exception of large hemangiomas or hemangiomatosis of skeletal muscle. These are aggressive, congenital lesions that are often unresectable because of extensive neurovascular and soft tissue involvement.136,137 Many of these extensive lesions result in amputations because of painful, dysvascular, or infected extremities. Most of these lesions are best diagnosed by open biopsy after MRI, CT scan with contrast, or arteriography. Well-localized lesions are more easily resected than the more extensive congenital lesions. Embolization and interferon treatment have had mixed results in halting the progression of disease.






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


