Chapter 8 The Structure and Derivation of Antibodies and Autoantibodies
The humoral immune response protects an organism from environmental pathogens by producing antibodies (immunoglobulins) that mediate the destruction or inactivation of microbial organisms and their toxins. To perform this function, the immune system generates antibodies to a diverse and changing array of foreign antigens, yet it must do so without generating pathogenic antibodies to self. The production of high-affinity antibodies that bind to self-determinants is a prominent feature of systemic lupus erythematosus (SLE).1 Some autoantibodies in SLE are considered markers for disease (anti-Sm/ribonucleoprotein [RNP], antinuclear antibody) because they have no established pathogenicity; others play a role in disease pathogenesis and cause tissue damage (anti-DNA, anticardiolipin, anti-Ro).2–6
Extensive investigations of autoantibodies in SLE have addressed the following specific questions:
1. Do polymorphisms of immunoglobulin variable region genes contribute to disease susceptibility?
2. Do B cells producing autoantibodies arise from an antigen-triggered and antigen-selected response? If so, are these triggering and selecting antigens self or foreign?
3. Are particular B-cell lineages or differentiation pathways responsible for autoantibody production?
4. What are the characteristics of pathogenic autoantibodies, and how do they mediate pathology?
5. What defects in immune regulation permit the sustained expression of pathogenic autoantibodies?
Structure of the Antibody Molecule
Antibodies are glycoproteins produced by B lymphocytes in both membrane-bound and secreted forms. They are composed of two heavy chains and two light chains. In general, the two heavy chains are linked by disulfide bonds, and each heavy chain is linked to a light chain by a disulfide bond. The intact molecule has two functional regions: a constant region that determines its effector functions and a variable region that is involved in antigen binding and is unique to a given B-cell clone (Figure 8-1).7 The light chains appear to contribute solely to antigen binding and are not known to mediate any other antibody function. In contrast, the heavy chains possess a constant region that determines the isotype (i.e., class: immunoglobulin M [IgM], IgD, IgG, IgA, or IgE) of the antibody molecule (Figure 8-2). Rarely, the same variable region associated with a different constant region may display an altered binding to antigen.8,9 IgM is the first isotype produced by a B cell and the first to appear in the serum response to a newly encountered antigen. IgM antibodies normally polymerize into pentamers known as macroglobulin, thus conferring higher functional binding strength, or avidity. A 15-kd glycoprotein called the J chain is covalently associated with the pentameric IgM and mediates the polymerization process.10,11 IgM antibodies can activate complement through the classical pathway and therefore cause lysis of cells expressing target antigens. Under the appropriate conditions, B cells producing IgM can switch to the production of the other isotypes. IgG is the predominant isotype of the secondary (also called memory) immune response. In humans, the IgG isotype is divided into four subclasses, IgG1, IgG2, IgG3, and IgG4, all of which possess different functional attributes. IgG1 is the most abundant in the serum. Antinuclear antibodies in SLE are mainly of IgG1 and IgG3 subclasses.12 In addition to activating complement, IgG antibodies can promote Fc receptor (FcR)–mediated phagocytosis of antigen-antibody complexes. High concentrations of antigen-IgG complexes can downregulate an immune response by cross-linking membrane immunoglobulin and the receptor FcRII on antigen-specific B cells. This may be an important mechanism for turning off antibody production after all the available antigen is bound to antibody, and there is some evidence for defective FcRII function in some patients with lupus. The IgA constant region allows antibody translocation across epithelial cells into mucosal sites such as saliva, lung, intestine, and the genitourinary tract; IgA antibodies can be found as monomers in serum and as dimers in the mucous secretions. The J chain, implicated in IgM polymerization, is not required for IgA dimerization but does have a role in maintaining IgA dimer stability and is essential for transport of IgA by the hepatic polymeric Ig receptor.13 IgE antibodies can trigger mast cells and eosinophils, which are important cellular mediators of the immune response to extracellular parasites and cause allergic reactions.
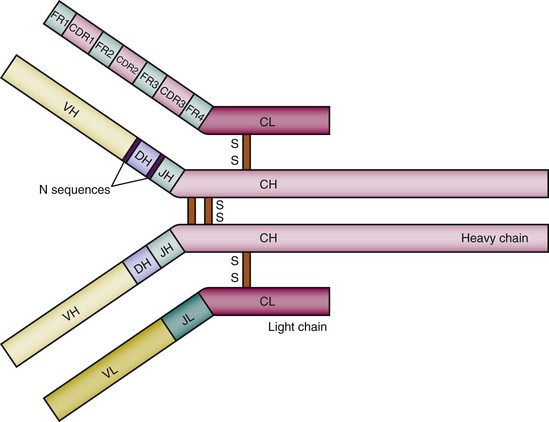

Every complete antibody has two identical antigen-binding sites, each of which is composed of the variable regions of a heavy and a light chain. Each variable region is divided into the highly polymorphic complementarity-determining regions (CDRs), and the more conserved framework regions (FRs). There are three distinct CDRs in both the heavy chain and the light chain, and the most variable portion of the antibody molecule is the CDR3.14,15 There are four FRs. When the variable regions from the light and heavy chain pair, hypervariable CDRs come together and generate a unique antigen-binding site (see Figure 8-1). X-ray crystallographic studies have shown that the amino acids of the CDRs are arranged in flexible loops but the FRs have a more rigid structure that maintains the spatial orientation of the antigen-binding pocket16—a finding consistent with the fact that CDRs contain the contact amino acids for antigen binding and thus contribute more than the FRs to antigenic specificity.
Antibody molecules can be cleaved into functionally distinct fragments by papain and pepsin.17,18 Limited digestion with papain cleaves the antibody into three fragments: two identical Fab (fragment antigen-binding) fragments and an Fc (fragment crystallizable) fragment. The Fab fragment consists of the entire light chain and the heavy-chain variable region with the CH1 domain. It contains the antigen-binding site, which is formed by the variable regions of the light and heavy chains. The Fc fragment is composed of the two carboxyterminal domains from the heavy chains, the hinge region, and CH2 and CH3, and interacts with soluble and cell membrane–bound effector molecules. The Fc fragment does not have antigen-binding activity. The Fab portions are linked to the Fc fragment at the hinge region, an arrangement that allows independent movement of the two Fabs.19 Another protease, pepsin, cleaves the antibody molecule on the carboxyterminal side of the heavy-chain disulfide bridges, producing several small fragments and an F(ab)2 fragment, which contains both Fabs linked to each other with an intact hinge region. F(ab)2 cannot be obtained from IgG2 by pepsin. However, lysyl endopeptidase digestion can generate F(ab)2 from IgG2.20 On the basis of the fact that the F(ab)2 fragment has the same avidity for antigen as the intact antibody but does not possess any effector functions, this cleavage product may have therapeutic applications.
The variable region of an antibody may itself serve as an antigen, called an idiotype. Antiidiotypes are antibodies that bind to specific determinants in the CDRs or FRs of other antibodies.21,22 Antibodies that have the same idiotype presumably have a high degree of structural homology and may be encoded by related variable region genes.23 Idiotypes have been postulated to be important in the regulation of the immune response because they can be recognized by both T and B cells.24–27 Antiidiotypic antibodies may therefore be useful reagents for tolerizing pathogenic autoantibody–producing B cells (see later).
Antibody Assembly
The immunoglobulin light-chain and heavy-chain variable region genes are formed by a process of rearrangement of distinct gene segments in B cells through a process called somatic recombination. During this process, V (variable), D (diversity), and J (joining) segments are brought together to form a heavy-chain variable region gene, and V and J segments to form a light-chain variable region gene.28–33
In humans, heavy-chain V, D, and J gene segments each come from gene clusters that are arrayed on chromosome 14 (see Figure 8-2).33,34 The 50 to 100 functional heavy-chain V segment genes are divided into seven families, which share 80% homology by DNA sequence primarily in FRs.35–38 V gene family members are interspersed along the V locus. There are approximately 30 functional D gene segments and six known J gene segments for the human immunoglobulin heavy chain.35
Assembly of the complete heavy-chain gene begins with the joining of a D segment from the D cluster to a J segment in the J cluster, mediated by DNA cleavage and deletion of the intervening DNA. In a similar manner, a V gene segment is next rearranged to the DJ unit to form a complete VDJ variable region.28,38 Each V, D, and J gene segment is flanked by conserved heptamer/nonamer consensus sequences known as recombination signal sequences (RSSs), which are crucial for the rearrangement process.35 This process of variable region recombination is very elaborate and requires a complex of enzymes called V(D)J recombinase.39 Most of these enzymes are also necessary for the maintenance of double-stranded DNA (dsDNA) and are present in all cells. However, for the first cleavage step, specialized enzyme products of the recombination-activating genes, RAG-1 and RAG-2, are required.40 The proteins encoded by these genes are active in the early stages of lymphoid development. Signals from both stromal cells and the cytokines interleukin-3 (IL-3), IL-6, and IL-7 are necessary for induction of RAG expression in lymphoid progenitors.41 RAGs initiate VDJ recombination by generating dsDNA breaks at the end of the RSS. Joining of the coding segments is mediated by the following enzymes involved in repair of dsDNA breaks: Ku70, Ku80, DNA-PKs, XRCC4, DNA ligase IV, Artemis, and Mre 11.42 Members of the high-mobility group family of proteins, HMG1 and HMG2,43 also play a significant role in the formation and stabilization of the precleavage and postcleavage synaptic complex.44,45
Antibody diversification can be further generated by the addition of P and N nucleotides at the VD and DJ junctions. If the single-stranded DNA (ssDNA) that is present after the break can form a hairpin loop, the resulting double-stranded (palindromic [P]) sequences are added at the junction. Alternatively, N-nucleotides, or non–template-encoded nucleotides, are randomly inserted at the VD and DJ junctions by the enzyme terminal deoxynucleotidyl transferase (TdT).46 Such N sequences are common in antibodies of the adult immunoglobulin repertoire but are less frequent early in the ontogeny of the B-cell repertoire.47 These random modifications create unique junctions and increase the diversity of the antibody repertoire. Because VDJ joining is imprecise and includes P and N sequences, CDRs of variable length and sequence are generated.
After generation of a functional heavy chain, the light-chain gene segments can rearrange from either of two loci, κ or λ. The ratio of the two types of light chains varies in different species. For example, in mice the κ/λ ratio is 20 : 1 and in humans it is 2 : 1. The light-chain isotype has in general not been found to influence major properties of the antibody molecule. The light-chain variable region is composed of only two gene segments: V and J. Genes for the V and J segments of κ light chains are located on chromosome 2 in humans. The κ locus contains approximately 40 functional V gene segments, which are grouped into seven families, and five J segments.48–52 The λ light-chain locus is on human chromosome 22 and contains at least seven V gene families with up to 70 members.53–57 As with the heavy chain, V and J elements of the light-chain loci also rearrange by recombination at heptamer/nonamer consensus sites. Only rarely are N sequences inserted at the VJ junction of the light chain.58
The importance of the V(D)J recombination process has been demonstrated in animal studies as well as in some hereditary immune disorders. Mutations that abolish V(D)J recombination cause an early block in lymphoid development resulting in severe combined immune deficiency (SCID) with a complete lack of circulating B and T lymphocytes. Mice missing either RAG-1 or RAG-2 are unable to rearrange immunoglobulin genes or T-cell receptor genes.59 In humans, a loss or marked reduction of V(D)J recombination activity can cause a T-B-SCID60,61 or B-SCID phenotype.62 Mutations that impair but do not completely abolish the function of RAG-1 or RAF-2 in humans result in Omenn syndrome, a form of combined immune deficiency characterized by lack of B cells and the presence of oligoclonal, activated T lymphocytes with a skewed T helper 2 (Th2) profile.63 It is clear, however, from studies of immunodeficient mouse strains that additional gene products are needed for successful rearrangement to occur. Defects in any of the components of the dsDNA break repair machinery, such as Ku70, Ku80, DNA PKs, DNA ligase IV, Artemis, and XRCC4, lead to an immunodeficient phenotype with increased radiation sensitivity as a common feature.64
Although the rearranged heavy-chain VDJ segment is initially joined with an IgM constant region gene, it can undergo a second kind of gene rearrangement during the secondary response to recombine with the other downstream constant region genes (see Figure 8-2).65–67 Switch sequences located upstream of each constant region gene mediate heavy-chain class switching.68
Although all somatic cells are endowed with two of each chromosome, only one rearranged heavy-chain gene and one rearranged light-chain gene normally are expressed by a B cell. This phenomenon is known as allelic exclusion. A productive rearrangement on one chromosome inhibits assembly of variable region genes on the other chromosome. Rearrangement of the first chromosome is often unproductive because of DNA reading frame shifts or because nonfunctional variable region gene segments called pseudogenes are used. If rearrangement on the first chromosome does not lead to the formation of a functional polypeptide chain, then the immunoglobulin genes on the other chromosome undergo rearrangement. Monoallelic expression avoids potential expression of immunoglobulins or B-cell receptors (BCRs) with two different specificities on the same B cell, which could interfere with normal selection processes (see later). Regulation of allelic exclusion seems to occur at the recombination level, as suggested by the observation that transgenic mice allow the expression of two prearranged alleles at either the heavy- or light-chain locus.69 Single-cell analysis of germline transcription in pro-B cells has shown transcription of Vκ genes on both chromosomes70; however, the earlier-expressed alleles are almost always the first to undergo rearrangement.71 Methylation of DNA mediates gene repression and, when found in the proximity to recombination sites, decreases the probability of recombination.72
Although the heavy chain has a single locus of V, D, and J segments on each chromosome, the light chain has two. The κ locus is the first set of light-chain gene segments to rearrange. If these rearrangements are nonproductive on both chromosomes, however, then the V and J segments of the λ locus rearrange to produce an intact light chain.73,74 Thus, the heavy chain has two loci from which to form a functional gene, but the light chain may rearrange at four loci. Moreover, additional or secondary rearrangements can occur in B cells already expressing an intact antibody molecule if that antibody has a forbidden autospecificity. These additional rearrangements, which are termed receptor editing, are important in allowing B cells to regulate autoreactivity.
Immune tolerance mediated by receptor editing occurs frequently in developing B cells.75 High-affinity receptor binding to self-antigen induces a new gene recombination76 and the replacement of the gene encoding a self-reactive receptor by a gene encoding a non–self-reactive receptor.77,78 Receptor editing occurs at both light- and heavy-chain loci, but at a much lower frequency at the heavy-chain locus.79 There is some debate about whether Ig gene rearrangement can occur also in mature B cells or only in immature B cells.80 RAG protein expression in germinal centers, as well as after immunization,81,82 has suggested that antibody genes may undergo modification not only in developing but also in mature B cells.82–84 Immunization of BALB/c mice with a multimeric form of a peptide mimotope of dsDNA induces the generation of dsDNA-reactive B cells. Mature B cells that respond to peptide reexpress RAG for a short time only, suggesting that receptor editing can also participate in peripheral tolerance.85 The regulation and function of secondary rearrangements of Ig genes in mature B cells remain incompletely understood, however, because some data suggest that rearrangement events can be initiated in germinal center B cells that fail to bind antigen.
Generation of Antibody Diversity
The immune system has several mechanisms to ensure a large antibody repertoire. Before exposure to antigen, B-cell diversity results from (1) combinations of V, D, and J gene segments and V and J segments into heavy- and light-chain genes, respectively, (2) junctional diversity produced by N or P sequence insertion and/or imprecise joining of gene segments, and (3) the random pairing of heavy and light chains. These three mechanisms are consequences of the process of recombination used to create complete Ig variable regions. The fourth mechanism, called somatic hypermutation (SHM), occurs later on rearranged DNA. This mechanism introduces point mutations into rearranged variable region genes (Box 8-1). These mechanisms are potentially capable of producing a repertoire of 1011 different antibodies.86
Box 8-1
Mechanisms of Antibody Diversity
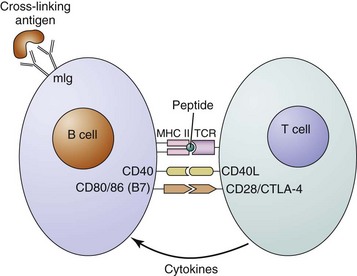
Cross-linking of surface immunoglobulin on the B cell by a multivalent antigen is the first in a series of critical steps that eventually can lead to B-cell activation and antibody production. After cross-linking of membrane immunoglobulin, the antigen-antibody complexes are internalized, and the antigen is cleaved and processed in the cell. Peptide fragments of protein antigen bound to the major histocompatibility complex (MHC) class II molecules are then expressed on the cell surface, where they can be recognized by antigen-specific helper T cells (Figure 8-3). These T cells provide the co-stimulation and cytokines that are necessary for full B-cell activation.
On initial exposure to an antigen, naïve B cells recognizing the antigen proliferate and begin to secrete IgM. These B cells can belong to the B1, marginal zone, subset or the follicular B cell subset. The antibodies of this primary immune response generally are polyreactive and display low affinity for a multitude of antigens, even for antigens without obvious structural homology (Table 8-1). The amplification of antigen-specific B follicular cells occurs in specific regions of the lymphoid tissue called germinal centers. Somatic hypermutation (discussed later), leads to the selection of high-avidity B-cell clones. Within the germinal center, heavy-chain isotype switching and further differentiation to plasma cells and memory B cells also occur.
TABLE 8-1 Distinguishing Features of the Naïve and Antigen-Activated Antibody Repertoire
| FEATURE | NAÏVE | ANTIGEN ACTIVATED |
|---|---|---|
| Isotype | Primarily immunoglobulin (Ig) M | Primarily IgG |
| Specificity | Polyreactive | Monospecific |
| Affinity | Low affinity | High affinity |
| Sequence | Germline gene encoded | Somatically mutated (high replacement–to–silent mutation [R:S] ratio) |
| Titer | Low titer | High titer |
Studies using mice with targeted disruptions of particular genes have shown that in addition to a cognate interaction between the T-cell receptor and an MHC class II molecule, other pairs of B cell–T cell contacts are necessary for germinal center formation and function (see Figure 8-3). One important interaction is between CD40 on the B cell and CD40 ligand (CD40L, gp39) expressed on activated CD4+ T cells. Activation of CD40 is thought to be necessary for the formation of germinal centers and germinal center reactions.87,88 Defective CD40L on T cells in humans and mice causes X-linked hyper-IgM syndrome type I, which is characterized by a defect in isotype switching and severe humoral immunodeficiency, leading to increased susceptibility to infections with extracellular bacteria.89 After the primary immune response is complete, specific antibody secretion decreases. Reexposure to the antigen and activated T cells, however, can activate memory B cells, which arise in the germinal center response to initiate the secondary immune response. The secondary serum response is characterized by rapidly produced high titers of IgG antibodies that have greater specificity and increased affinity for the antigen.90–92 The increase in both affinity and specificity is a consequence of SHM and the selection process within the germinal center. Anti-dsDNA antibodies, which are the well-characterized pathogenic autoantibodies to date, possess all the features of secondary response antibodies (see Table 8-1; see Chapter 23).93–96
Somatic Hypermutation
Somatic point mutations are single-nucleotide substitutions that can occur throughout the heavy- and light-chain variable region genes97–99; they represent a site-specific, differentiation stage–specific, and lineage-specific phenomenon.100 Somatic mutation takes place in dividing centroblasts (noncleaved cells in the centers of lymphoid follicles), in which rearranged Ig variable region genes undergo a mutation rate of 1 base pair (bp) per 103 cell divisions, compared with 1 bp per 1010 cell divisions in all other somatic cells. The DNA mismatch repair system has been implicated in Ig gene mutation because it functions generally to correct point mutations in DNA. A genetic deficiency in a component of the mismatch repair system, PMS2, has been shown to enhance the rate of mutation, suggesting that the DNA mismatch repair system may be altered in hypermutating B cells.101 Similarly, mice deficient in Msh6, a component of the mismatch repair system, have altered nucleotide targeting for mutations.102 Because somatic mutation occurs concurrently with heavy-chain class switching, although by a different mechanism, mutation is more common in IgG than in IgM antibodies.
The generation of high-affinity antibodies through B-cell maturation with SHM and class switch recombination (CSR) critically depends on the action of activation-induced cytidine deaminase (AID).103 AID is a member of a family of APOBEC (apolipoprotein B messenger RNA–editing, enzyme-catalytic, polypeptide-like 3G) cytidine deaminases that causes DNA conversions of cytosine to uracil, generating mutations in the immunoglobulin gene that can increase antibody affinity for the antigen.104 AID deficiency in humans causes a disorder called hyper-IgM syndrome type 2, which is characterized by elevated serum values of IgM and undetectable levels of IgG, IgA, and IgE.105 Mice with a homozygous deletion of AID display normal B-cell maturation but are deficient in SHM and CSR, whereas overexpression of AID is sufficient to induce SHM and CSR in B-cell lines or fibroblasts.106,107 AID expression is tightly regulated and appears to be restricted to GC, although clearly CSR can occur outside GC. Genomic instability and higher mutation rates are likely to occur in the presence of poorly regulated AID expression, possibly leading to malignancies.104
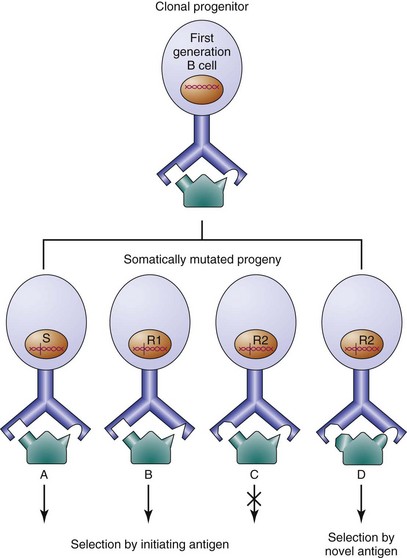
Genealogies of B cells with serial mutations in their immunoglobulin gene sequences demonstrate how point mutations can lead to antibodies with altered affinity for antigen (Figure 8-4).108–111 Although B cells producing antibodies with decreased affinity appear within the germinal center, progression of these cells to the plasma or memory cell compartment is rare, because they fail to expand further in the germinal center response. In contrast, B cells producing antibodies of higher affinity continue to expand. SHM is an important process in the generation of high-affinity antibodies, and a suboptimal frequency of Ig V gene mutation leads to common variable immunodeficiency (CVID).112 Mutated antibodies also can acquire novel antigenic specificities. In one in vitro system, a single amino acid change in a protective antipneumococcal antibody results in reduced binding to pneumococci and a newly acquired affinity for dsDNA.113 Abundant evidence suggests that antibodies to foreign antigen also can acquire autospecificity in vivo through somatic point mutation.114,115
Because a given amino acid can be encoded by more than one DNA triplet, not every point mutation causes an amino acid substitution that can change antibody affinity for an antigen. It is possible to indirectly analyze antigen selection during the course of the germinal center response by calculating the ratio of replacement (R) mutations (mutations that lead to amino acid changes) to silent (S) mutations (mutations that do not lead to such changes) in rearranged antibody genes. Purely random point mutations within a DNA sequence containing equal numbers of each possible codon would result in a predicted random R : S ratio of approximately 3 : 1.116,117 The random R : S ratio for a particular DNA sequence, however, might be lower or higher, depending on the actual codon usage.118,119
In an antigen-selected response, one might expect a higher than random R : S ratio, because B cells containing mutations leading to higher affinity for antigen would be favored to proliferate. Further, antigen selection would predict a higher frequency of R mutations in the CDRs, because these regions include the contact amino acids for antigen binding. This type of analysis has been performed to assess whether certain autoantibodies arise from antigen selected responses.94–96 There are two concerns, however, with this analysis. First, the assumption of purely random mutation is incorrect; studies have now shown that bias for particular kinds of mutations occurs and that hot spots of mutation exist.120 Second, although antibodies with a higher-than-random R : S ratio probably are part of an antigen-selected repertoire, the converse clearly is not true; a single amino acid substitution is capable of conferring a tenfold increase in affinity.121,122 Thus, antigen selection may occur in the absence of a high R : S ratio.
B-Cell Subsets: Implications for SLE
B-1 cells (also CD5 or Ly-1) represent a distinct population of B cells.123,124 B-1 cells are the only subset of B lymphocytes that constitutively express the pan–T-cell surface antigen CD5. B-1 cells are mainly found in the peritoneal and pleural cavities of mice (accounting for 35% to 70% of total B cells found in these sites) and are rare in lymphoid organs and blood.125 They can be further divided into B-1a and B-1b cells and are usually recognized as having the following surface phenotype: CD19hiCD23−D43+gMhiIgDvariableCD5±. Data showing that CD5 is implicated in the maintenance of tolerance in anergic B cells,126 along with data demonstrating that CD5 mediates negative regulation of BCR signaling in B-1 cells,127 support the hypothesis that the expression of CD5 may help inhibit autoimmune responses. The phenotype of B-1 cells in humans has been reported to be CD27+, CD43+, CD70−; this subset contains DNA-reactive B cells.128
A two-pathway model of B-1 cell development has been proposed on the basis of the identification of a bone marrow and fetal liver precursor to mainly B-1b cells and the observation that B-1a cells can be differentiated from B-2 cell precursors under certain physiologic conditions.125 B-1 cells are unique among mature B lymphocytes in that they appear to be a self-replenishing population that arises in the fetal liver.129 Being a major source of natural autoantibodies,130–132 the B-1 lineage is of particular interest to those studying autoimmunity. Elevated numbers of B-1 cells are present in the autoimmune New Zealand black (NZB) mouse strain,129 and prevention of the autoimmune symptoms has been reported with their elimination.133 B-1 cell expansion is found in some patients with rheumatoid arthritis and Sjögren’ syndrome,134 but an association with SLE is weaker.135,136
B-1 cells generally express germline-encoded, polyreactive IgM antibodies with limited V gene segment usage.129–131 Much controversy exists about the physiologic function of the B-1 lymphocytes, although there is now growing evidence that many of the low-affinity autoantibodies made by this B-cell subset are important in the clearance of apoptotic debris. Adoptive transfer experiments have shown that B-1 cells are poor at forming germinal centers,137 which are characteristic of a T-dependent B-cell response and are thought to be necessary for antigen selection and class switching; however, class-switched, somatically mutated B-1 antibodies that appear to show evidence of antigen selection have been isolated from humans.138
MZ (marginal zone) B cells share many features with B-1 B cells. They are phenotypically characterized by cell surface expression of IgMhiIgDloCD21hiCD22hiCD23loCD1hi and reside in the marginal zones that girdle the follicles in spleen and tonsils.139 Their hallmark functional characteristic is represented by early activation and rapid Ig secretion in response to T-independent (TI) antigens, which arrive via a hematogenous route in the spleen. Like B-1 cells, MZ B cells are key players of innate immunity because they respond rapidly to antigen and do not generate a memory response. Although it has been generally accepted that MZ B cells are a self-renewing and mostly nonrecirculating population,140,141 later studies suggest that a large population of IgM-positive peripheral B cells correspond to circulating splenic MZ B cells.142,143
Both MZ and B-1 B cells have a high antigen presentation capacity and are strategically located to encounter and process foreign antigens. Both cells secrete polyreactive “natural” antibodies, including self-reactive ones that are generally germline encoded.144 Low titers of low-affinity autoantibodies are part of the normal B-cell repertoire.145–148 Such antibodies are not unique to any autoimmune disease, nor is there any evidence that they are pathogenic. These natural autoantibodies resemble the antibodies of a primary immune response, in that they are mainly IgM and polyreactive and bind to a wide variety of both autoantigens and foreign antigens that often have no apparent structural homology.149–151 “Natural” antibodies have also been shown to bind to altered phospholipids expressed on the surfaces of cells undergoing apoptosis. The opsonization of apoptotic cells increases their clearance and routes them to nonimmunogenic pathways.152 Although sequence analysis shows that the antibodies made by MZ B cells are encoded mainly by germline (i.e., unmutated) genes,153–157 numerous exceptions exist.158 Analysis of the variable regions of natural autoantibodies suggests that they may contain more flexible hydrophilic amino acid residues in their CDRs than somatically mutated, affinity-matured antibodies, as well as longer CDRs,158 features that may explain their polyreactivity. It is thought that they present a shallow groove for antigen binding that can accommodate more diverse structures.
There are some indications that the B cells producing natural antibodies may be clonally related to pathogenic B cells. Idiotypic analyses of natural anti-DNA antibodies from normal individuals and of potentially pathogenic anti-DNA antibodies from patients with SLE demonstrate that cross-reactive idiotypes are present in both populations.159,160 Some investigators have speculated that natural autoantibodies can be the precursors to pathogenic autoantibodies,161,162 and other data suggest that the two classes of autoantibodies arise from distinct B-cell populations and that the SLE autoantibodies arise by the somatic mutation of genes that encode protective antibodies.93,163–168 Adoptive transfer experiments of MZ B cells, like those of B-1 cells, have demonstrated T-dependent class-switching and SHM, resulting in the production of high-affinity antibodies.169,170 If one assumes that MZ B cells can undergo affinity maturation, it is conceivable that an enhanced differentiation of MZ B cells along this pathway could contribute to autoimmunity. Another potential role for MZ B cells in autoimmunity is as antigen-presenting cells for self-antigens, resulting in the activation of autoreactive CD4+ T cells. These T cells can then amplify an autoreactive B-cell response by activating additional autoreactive B cells.
Understanding of innate immune B cells in humans has been further advanced through the study of a population of B cells that can be identified using a monoclonal antibody (9G4) that binds to a unique epitope encoded by the human heavy-chain variable region gene V4-34.171 These 9G4-positive B cells represent 5% to 10% of the mature naïve B-cell repertoire and recognize autoantigens and pathogens. In addition, these cells are present in the MZ B cell compartment and are normally excluded from the T-dependent IgG memory repertoire. However, in patients with SLE, 9G4-positive B cells are expanded in the IgG memory population, supporting the hypothesis that inappropriate positive selection of innate B cells into an adaptive immune phenotype is a feature of autoimmunity. Although 9G4-positive antibodies have not been demonstrated to have a direct pathogenic effect, they are elevated in up to 75% of patients with active SLE.
Follicular B cells have the most diverse immunoglobulin repertoire. These are the B cells that participate in T-cell–dependent antibody responses. Follicular B cells, when they encounter antigen and T-cell help, can become short-lived plasma cells or can enter into a germinal center response in which long-lived plasma cells and memory cells are generated. Because the recognition of an increased expression of type I interferon–inducible genes, and an interferon signature in mononuclear cells of patients with SLE, several investigators have studied a mouse model of SLE in which disease is accelerated through the administration of type I interferon. Interestingly, this interferon-accelerated model is characterized by the presence of short-lived plasma cells as opposed to germinal center–matured cells,172 perhaps related to interferon induction of IL-12.173 During the germinal center response, heavy-chain class switching and SHM of Ig variable region genes occur. The process of SHM can clearly generate autoreactivity. Studies of both MRL-lpr/lpr and NZB/W mice have shown that many anti-DNA antibodies display extensive somatic mutation, which is responsible in some cases for increasing affinity for DNA and in other cases for the acquisition of autoreactivity. In these models there are clearly impairments in both central tolerance and peripheral tolerance, with defects in negative selection of antigen-naïve and antigen-activated B cells, respectively. There are now a number of mouse models of SLE in which all the DNA-reactive B cells appear to be generated in the germinal center response, through the process of SHM.174 These models are of particular interest because B-cell autoreactivity appears to be regulated appropriately at early stages of B-cell development but not in germinal center B cells.
Toll-Like Receptors in B-Cell Function
B cells express Toll-like receptors (TLRs), which recognize specific molecular determinants common to many pathogens. In mouse B cells, coengagement of TLRs and the BCR acts synergistically to induce activation; in humans, TLR expression appears to be induced following BCR activation.175
TLRs have been shown to bind exogenous ligands, such as lipopolysaccharides (LPSs), single- and double-stranded RNA and dsDNA derived from bacteria, and neutrophils undergoing NETosis, or from apoptotic debris.176–178 Inducible TLR expression and B-cell activation from a wide range of self-ligands and foreign ligands may provide a link between innate immune dysregulation and autoimmunity. Interestingly TLR-dependent activation of B cells expressing antichromatin antibodies leads to isotype switching and SHM in the absence of T-cell co-stimulation.179 A number of additional factors have been found to promote T-independent isotype switching, including B-cell–activating factor (BAFF) and type I IFN. In addition, CpG binding to TLR9 in B cells from several lupus mouse strains increases the secretion of IL-10 and results in the suppression of IL-12 production.180 IL-10 has been shown to be elevated in patients with SLE, and serum levels can correlate with disease activity.181,182 In an uncontrolled study, a small number of patients with active SLE were given anti–IL-10 antibody and experienced an improvement of disease activity.183 Similarly, anti–IL-10 treatment of NZB/W mice resulted in delayed onset of lupus-like disease.184 However, MRL-Fas(lpr) IL-10−/− mice showed an increased severity of lupus and higher concentrations of anti-dsDNA antibodies.185 An IL-10–producing B-cell subset (regulatory B cells [Bregs]) has now been identified that can suppress immune responses to foreign antigen and self-antigen.186 Transitional (CD19+CD21hiCD23hiCD1dhi) are able to suppress mouse models of inflammatory arthritis, experimental allergic encephalitis, and lupus in an IL-10–dependent fashion.187 A better understanding of the impact of Bregs in lupus may lead to new therapeutic targets.
Pathogenic Autoantibodies
Indirect evidence for the pathogenicity of several autoantibodies present in SLE includes their association with clinical manifestations in SLE and their presence in affected tissue. There is growing evidence to directly support the pathogenic potential of several lupus-associated autoantibodies. Glomerulonephritis has been shown to develop in a transgenic mouse expressing the heavy and light chains of the secreted form of an anti-DNA antibody, thereby confirming that anti-DNA antibodies cause renal disease.188 Support for the pathogenic role of anti-DNA antibodies in nephritis can also be found in autoimmune disease models displaying high titers of anti-DNA antibodies together with immunoglobulin deposition in the kidney and histologic nephritis.189–192 Perfusion of monoclonal mouse and polyclonal human IgG anti-DNA antibodies through isolated rat kidney induces significant proteinuria and decreased clearance of inulin.193 Addition of plasma as a source of complement markedly increases proteinuria, whereas preincubation of the antibodies with DNA can abolish binding to renal tissue.193 It is still unknown, however, whether pathogenic anti-DNA antibodies form immune complexes with antigen in situ or the antibodies bind to a target antigen that is actually some component of glomerular tissue and/or tubular components. A decrease in binding of anti-DNA antibodies to glomerular elements with DNase treatment occurred in some experimental models194 but not in others,195 suggesting that both models pertain; some anti-DNA antibodies directly cross-react with glomerular antigens, whereas other anti-DNA antibodies may bind via a DNA-containing bridge. A number of investigators have administered monoclonal anti-DNA antibodies to nonautoimmune mice, either intraperitoneally in the form of ascites-producing hybridomas or intravenously as purified immunoglobulins.196,197 In these models, it is possible to demonstrate that anti-DNA antibodies differ with respect to pathogenicity,197,198 with some antibodies depositing in the kidney and others not. Moreover, those antibodies that are deposited in the kidney may differ with respect to the localization of deposition. In studies performed with the congenic mouse strain NZM2328.C57Lc4, chronic glomerulonephritis and severe proteinuria develop despite the fact that the mice do not generate autoantibodies to dsDNA or other nuclear antigens,199 consistent with the clinical observation that kidney disease can arise in individuals with no DNA-reactive antibodies.
Studies have also elegantly demonstrated the arrhythmogenic potential of anti-Ro antibodies. Affinity-purified anti-Ro antibodies from mothers with lupus whose babies have congenital heart block have been reported to inhibit calcium currents and induce complete heart block in an ex vivo perfused human fetal heart system.200 In another study, immunization of female BALB/c mice with recombinant La and Ro particles led to first-degree atrioventricular block in 6 of 20 pups born to immunized mothers and rarely to more advanced conduction defects.201 Finally, passive transfer of purified human IgG containing anti-Ro and anti-La antibodies to pregnant BALB/c mice was found to result in fetal bradycardia and first-degree atrioventricular block.202
Experimental evidence also supports the close epidemiologic association between antiphospholipid antibodies and thrombosis. Following experimental induction of vascular injury in mice, injection of affinity-purified immunoglobulin from patients with antiphospholipid syndrome was found to result in a significant increase in thrombus size and a delay in disappearance of the thrombus.203 Injecting human monoclonal anticardiolipin antibodies into pregnant BALB/c mice was reported to lead to fetal resorption and a significant decrease in placental and fetal weight.204 Similar results have been obtained with passive transfer of monoclonal murine and polyclonal human anticardiolipin antibodies.205
Heavy-chain isotype appears to be important in determining the pathogenicity of autoantibodies. For example, marked differences in the severity of induced hemolysis exist among IgG isotype switch variants of an antierythrocytic antibody that are related to the capacity of each isotype to bind to Fc receptors.206 In murine lupus, the switch from serum IgM anti-DNA activity to IgG anti-DNA activity heralds the onset of renal disease.207 Similarly, human IgG antibodies present in the immune complex deposits within the kidneys of patients with SLE appear to trigger mesangial cell proliferation and subsequent tissue damage to a greater extent than IgM antibodies, perhaps because mesangial cells or infiltrating mononuclear cells have Fc receptors for IgG.208 The importance of isotype for anticardiolipin antibodies is intriguing2; several groups have noted that IgG antiphospholipid and beta 2–glycoprotein antibodies correlate better with clinical thrombosis than other isotypes do (see Chapter 27). Nevertheless, pathogenicity has been shown also for IgM and IgA antibodies.203 IgM and IgA anticardiolipin antibodies also correlate with specific disease phenotypes. For example, IgM antiphospholipid antibodies are associated with hemolytic anemia.209
It was formerly widely believed that antibodies could not penetrate live cells and that nuclear staining of sectioned tissues was an artifact of tissue preparation. There is now evidence that some anti-DNA and anti–ribosomal P autoantibodies bind to the cell surface, traverse the cytoplasm, and reach the nucleus. Furthermore, data demonstrate a pathogenic effect from cellular penetration by autoantibodies.210–212 Although antigen translocation to the cell membrane may explain the accessibility of normally intranuclear antigens to interaction with autoantibodies,213,214 the capability to penetrate live cells and interact with cytoplasmic or nuclear components may be an additional pathogenic characteristic of some autoantibodies.
This chapter discusses aspects of autoantibody production, but it is increasingly evident that autoantibody-mediated tissue damage requires not just the presence of autoantibodies with particular pathogenic features but also the display of a specific antigen in the target organ.215 Differential display of antigen at the level of the target organ may contribute to genetic susceptibility to autoimmune disease. Evidence for such a hypothesis comes, in part, from a murine model of autoimmune myocarditis, in which differential susceptibility to antimyosin antibody–induced disease in different mouse strains depends on differences in the composition of cardiac extracellular matrix.216 Similarly, in a rat model for tubular nephritis, antibody-mediated disease depends on genetically determined antigen display in the renal tubules.217
Genetic and Molecular Analysis of Anti-DNA Antibodies
Genetic analyses of anti-DNA antibodies in both human and murine lupus have provided important information regarding the production of autoantibodies. There is currently no evidence that a distinct set of disease-associated, autoreactive V region genes is present only in individuals with a familial susceptibility to autoimmunity and is used to encode the autoantibodies of autoimmune disease. It is also clear that no particular Ig V region genes are absolutely required for the production of autoantibodies (reviewed in reference 218). Immunoglobulin genes that are present in a nonautoimmune animal clearly are capable of forming pathogenic autoantibodies. The offspring of a nonautoimmune SWR mouse and an NZB mouse (SNF1 mice) spontaneously produce autoantibodies,219 with a large percentage of the anti-DNA antibodies that are deposited in the kidneys of SNF1 mice having been encoded by Ig genes derived from the nonautoimmune SWR parent.219 In fact, both idiotypic and molecular studies show that the V region genes used to produce autoantibodies in lupus are also used in a protective antibody response in nonautoimmune individuals.220,221 Autoantibodies bear cross-reactive idiotypes that also are present on the antibodies that are made in response to foreign antigens, and V region genes used to encode autoantibodies also encode antibodies to foreign antigen.222–225 Indeed, a number of autoantibodies cross-react with foreign antigens, demonstrating that the same V region gene segments can be used in both protective and potentially pathogenic responses.226–228 These cross-reactive antibodies are capable of binding to bacterial antigen with high affinity, but they also possess specificity for a self-antigen. Patients with Klebsiella infections and individuals vaccinated with pneumococcal polysaccharide develop antibacterial antibodies expressing anti-DNA cross-reactive idiotypes.220,229 In vivo, cross-reactive antibodies with specificity to both pneumococcus and dsDNA are protective in mice against an otherwise lethal bacterial infection, yet they also can deposit in the kidney and cause glomerular damage.230 It appears that cross-reactive antibodies are routinely generated during the course of the normal immune response in the nonautoimmune individual. Ordinarily, however, autoreactive B cells expressing a self-specificity are actively downregulated and contribute little to the expressed antibody repertoire.114
Although there is no evidence that specific genes encode only autoantibodies, some data suggest that autoantibodies are encoded by a somewhat restricted number of immunoglobulin V region genes.231–233 In murine lupus, extensive analyses of anti-DNA–producing B cells show that 15 to 20 heavy-chain V region genes encode most anti-DNA antibodies.165,234–236 One study found a dramatic increase in the frequency of use of a particular J558 heavy-chain gene in autoimmune than in normal mice, whereas nonautoimmune mice that were immunized with an immunogenic DNA/DNA-binding peptide complex displayed intermediate usage.233 This finding supports the concept that differences in V gene usage that may be seen between autoimmune and nonautoimmune mice are quantitative rather than reflecting a true qualitative difference. Although molecular studies of human antibodies are more limited, idiotypic analyses also suggest restricted V gene usage. This observation is important because it suggests that antiidiotypes can play a role in therapeutic strategies. Furthermore, analysis of restriction fragment length polymorphisms, which is a tool used to identify the similarities and differences among particular genes in a population, has been used to examine whether distinct Ig gene polymorphisms are associated with SLE.237–239 A deletion of a specific heavy-chain V gene, hv-3, was reported to be more frequent in individuals with SLE or rheumatoid arthritis.240,241 A specific germline Vκ gene, A30, was found to increase the cationicity (and therefore the pathogenicity) of human anti-DNA antibodies. A defective A30 gene was found in eight of nine patients with lupus without nephritis, but this gene was normal in all nine patients with lupus with nephritis.242 Polymorphism at the Vκ gene locus may then contribute to susceptibility to lupus nephritis. Although these studies look at only small numbers of patients, they suggest that polymorphisms in immunoglobulin genes may make some contribution to the generation of autoantibodies and expression of human lupus. Nevertheless, the anti-DNA response is no more restricted than are many responses to foreign antigen, and the restricted V region gene usage does not appear to be skewed toward particular gene families.
SHM is one mechanism by which protective, antiforeign antibodies may evolve into pathogenic autoantibodies (see Figure 8-4).243,244 The characteristics and mechanics of SHM in SLE are, therefore, of interest. Examining ten human antibodies positive for a specific, lupus-associated idiotype (F4), Manheimer-Lory245 found no change in the frequency of somatic mutations or the distributions of such mutations in CDRs. Although the normal process of somatic mutation is generally random, there is some bias for mutation at specific sequence motifs, termed mutational “hot spots.” Surprisingly, F4-positive antibodies displayed abnormal somatic mutation, as shown by a decrease in hot-spot targeting. Mice transgenic for the antiapoptotic gene bcl-2 also display this decreased targeting of mutations to hot spots,246 so the decreased targeting in F4-positive antibodies derived from patients with lupus may reflect an abnormal process of B-cell selection rather than defective machinery for somatic mutation. Studies have been performed on the mutational process in the V gene repertoire in individual B cells from a small number of patients with lupus.247 The frequency of mutations was increased in both productive and unproductive Vκ rearrangements, with evidence of increased targeting to mutational hot spots in framework regions, consistent with altered selection. A single study in mice found essentially no differences in somatic mutation between B cells of an autoreactive strain and those of a normal strain.248 Conflicting data prevent drawing firm conclusions as yet.
Autoantibody Induction
Autoantibodies that are present in SLE may be germline-encoded or may reflect the process of SHM,95,249 suggesting exposure to antigen and T-cell help. For some autoantibodies, mutation of the germline sequences clearly is crucial in generating the autoantigenic specificity.95 These antibodies have a high R : S ratio, primarily in CDRs; however, the pitfalls of R : S ratio calculations have been discussed and should be considered in the analysis of anti-DNA antibodies.120,121 There also are lupus autoantibodies that have a high R : S ratio in framework regions.250 Because these framework region mutations are less likely to alter antigenic specificity, it is tempting to speculate that they instead may facilitate escape from a putative regulatory mechanism.
There are various hypotheses regarding the nature of an eliciting antigen or antigens in SLE (Box 8-2). Several lines of evidence support the role of foreign microbial antigens in the generation of autoantibodies.251 Lupus-prone strains of mice carrying the xid mutation, which impairs production of the antipolysaccharide antibodies that are required for antibacterial immunity, demonstrate much lower titers of anti-DNA antibodies and decreased renal disease.252 Similarly, autoimmune-prone NZB mice raised in a germ-free environment produce reduced titers of anti-DNA antibodies and show delayed onset of autoimmune manifestations.253 It has been shown that raising lupus-prone lymphoproliferative (MRL/lpr/lpr) mice in a germ-free environment and feeding them a filtered, antigen-free diet significantly decreases the severity of renal disease.254 Evidence that an antipneumococcal antibody can spontaneously mutate to become an anti-DNA antibody in an in vitro system,113 as well as in response to immunization with a pneumococcal antigen in vivo,114 also supports a close structural relationship between the autoantibody response and a protective antibacterial response. Finally, to further demonstrate the close relationship between a protective antibacterial and autoantibody response in lupus, Kowal255 generated a combinatorial immunoglobulin expression library in phage from splenocytes of a patient with lupus who was immunized with pneumococcal polysaccharide. Four of eight of the monovalent Fab fragments selected for expression of an SLE-associated idiotype bound both pneumococcal polysaccharide and dsDNA, indicating that a significant portion of the human antipneumococcal response in SLE is cross-reactive with self-antigen.52









