Overview.
Achondroplasia, an abnormality of endochondral ossification, is the most common form of skeletal dysplasia and occurs in approximately 1 of 25,000 live births (
14,
15).
Achondroplasia is caused by a gain of function in the mutation of a gene that encodes for fibroblast growth factor receptor 3 (
FGFR3) (
16,
17,
18 and
19). Achondroplasia arises from a point mutation on the short arm of chromosome 4 at nucleotide 1138 of the
FGFR3 gene. The mutation is located on the distal short arm of chromosome 4. The result of this mutation is endochondral-ossification-engendered underdevelopment and shortening of the long bones that does not involve intramembranous or periosteal components.
Achondroplasia is inherited as a fully penetrant autosomal dominant trait, but more than 80% of such cases are sporadic, meaning both parents are unaffected (
20). If one of the parents is affected, there is a 50/50 chance that the child will develop achondroplasia (
14,
15,
20). However, because there is also an increased incidence when the parents are more than 33 years old at the time of conception, a
de novo mutation is implied (
21).
Etiology.
The cause of achondroplasia is a single-point mutation in the gene that encodes for
FGFR3. FGFR3 mutations have also been found in individuals with thanatophoric dysplasia and hypochondroplasia. Almost all people with achondroplasia have the same recurrent
G-380Rlocus mutation, which causes a change in a single amino acid. This mutation substitutes an arginine for a glycine residue in the transmembrane domain of the tyrosine-coupled transmembrane receptor in the physis (
1,
22).
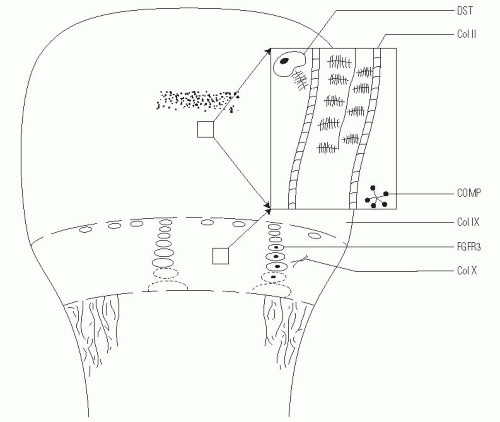
FGFR3 is expressed in the cartilaginous precursors of bone, where it is believed to decrease chondrocyte proliferation in the proliferative zone of the physis and to regulate growth by limiting endochondral ossification (
23). However, in persons with achondroplasia, articular cartilage formation, articular cartilage development, and the intramembranous and periosteal ossification processes are unaffected (
24).
It is not known why the proximal portions of the long bones (rhizomelic) are affected more than the distal aspects.
Clinical Features.
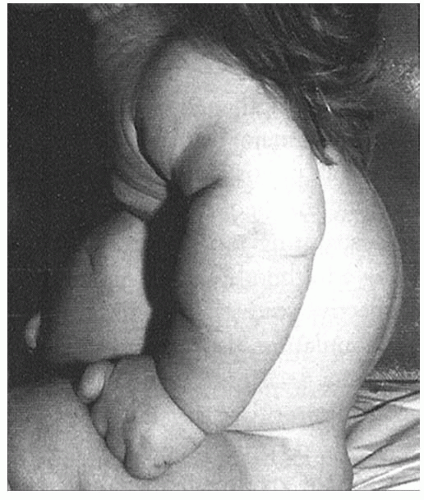
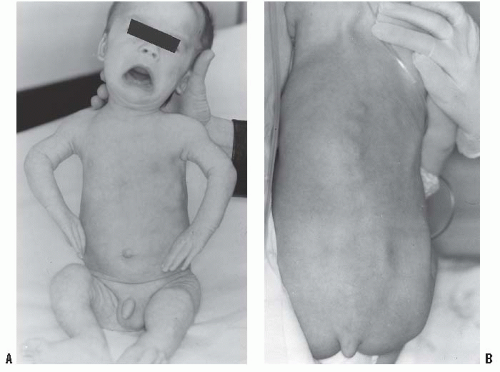
In the achondroplastic population, the extremities are most affected, that is, they are shorter than those in an unaffected individual. The most commonly affected bones are the humerus and femur, which present a rhizomelic appearance (
25). The trunk length is within normal limits or at the lower end of normal limits. This combination typically results in the fingertips reaching only to the tops of the greater trochanter (
26), a condition that can lead to possible difficulties in personal hygiene and care and that can worsen as decreasing amount of flexibility occurs in the normal aging process (
Fig. 7-2).
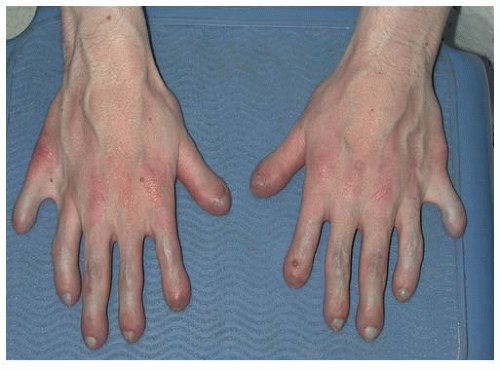
The hands are described as trident in nature, that is, the individual is unable to oppose the third and the fourth ray, leaving a space that cannot be closed. There are flexion contractures at the elbow and decreased ability to supinate, most often secondary to the fact that the radial head can be subluxed, as evidenced on radiographs. None of the above features are clinically important, but a nonknowledgeable physician might misdiagnose such a presentation as a “nursemaid’s elbow” and incorrectly attempt a reduction.
The facial appearance of patients with achondroplasia is characterized by an enlarged head, mandibular protrusion, frontal bossing (flattened or depressed nasal bridge), and midface hypoplasia. The bones in the midface are more affected than the other facial bones because of their endochondral origin (
16).
Although the lower extremities are typically in varus secondary to knee and ankle morphology, the lower extremities can be straight or occasionally in valgus, and internal tibial torsion may also be seen. Typically, the knee and ankle joints have excessive laxity, although usually such patients do not develop premature arthritis. The femoral necks are often shortened, giving an appearance of coxa breva.
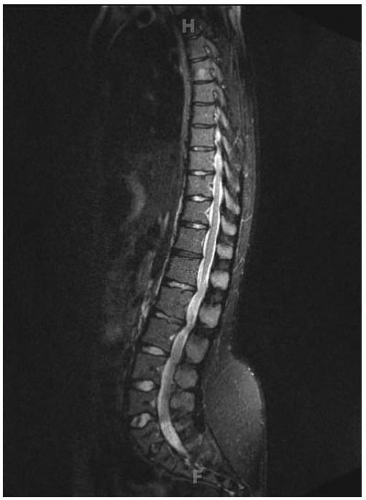
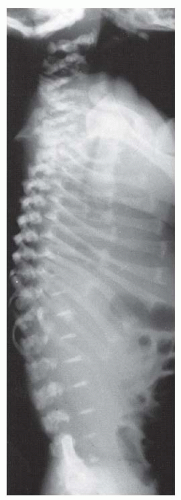
In terms of the spine, kyphosis at the thoracolumbar junction is very common and is typically seen in the first 1 to 2 years of life (
Fig. 7-3). In most children, this condition will correct spontaneously within a few months of ambulation, although ambulation is often delayed in patients with achondroplasia (
27). As kyphosis improves, lumbosacral lordosis may seem to progress.
One study found that life expectancy is not substantially diminished in individuals with achondroplasia (
28), but a more recent report has indicated a higher mortality rate in 30- to 50-year-old people with achondroplasia compared with age-matched controls (
29). This reported increased mortality is typically secondary to heart disease. One indicator for heart disease is abnormal blood pressure, but it is possible that standard blood pressure cuffs may underestimate the pressures in the achondroplastic population, leading to the nonidentification and nontreatment of a large number of patients with high blood pressure. New cuffs have been developed and their use for this population is being reviewed.
Growth and Development.
In most children with achondroplasia, growth and development fall behind those of unaffected children.
Widely available growth charts (
25) indicate that the infant with achondroplasia is shorter than an unaffected infant, a
height deficit that increases markedly during the first few years of life and becomes even more marked during the growth spurt at puberty (
30). The average height for an adult with achondroplasia is 132 cm for men and 125 cm for women (
20).
Children with achondroplasia also have delayed motor milestones (head control, 4 months; sitting up independently, 10 months; ambulation, 18 to 20 months) (
31,
32), and threequarters of them have ventriculomegaly (
33).
Historically, hydrocephalus was thought to be the cause, leading also to macrocephaly, but only a very small subset has been shown to have clinically significant hydrocephalus (
34); standardized head circumference charts can help track such children (
35). Ventricular peroneal shunting is indicated only for rapid progression of head circumference, or for signs and symptoms of increased intracranial pressure.
Mental development is typically normal in children with achondroplasia, but physical manifestations are often delayed, especially in the first 2 to 3 years of life (
36). Typically, motor development normalizes by 3 years of age. There are standardized developmental charts that are available for monitoring such children.
Foramen magnum stenosis is one of the earliest serious health consequences faced by some children with achondroplasia. Its symptoms, which most commonly occur in the first 2 years of life but which may present later (
37), include chronic brain stem compression, sleep apnea, lower cranial nerve dysfunction, difficulty in swallowing, hyperreflexia, hypotonia, weakness, paresis or clonus, and severe developmental delay, and are quantified in sleep studies (
34,
38,
39,
40,
41 and
42). The most common presenting symptom of foramen magnum stenosis is respiratory difficulty with excessive snoring or apnea (
43). Apnea can be central in nature (because of brainstem compression) or just obstructive because of the individual’s small midface. The American Academy of Pediatrics recommends screening for foramen magnum stenosis with polysonography and computed tomography (CT) or MRI in all infants with achondroplasia. Because CT and MRI in the first year of life require sedation, the child who is developing well, has no abnormal reflexes, and is alert, oriented, and meeting all milestones can typically just be followed clinically. If head circumference changes or if a patient is not reaching milestones, a sleep study should be ordered. If the sleep study is abnormal, then a CT or an MRI scan should be obtained. We prefer MRI because, in our opinion, it produces a better image of the brain stem and the upper cervical spinal cord.
Some studies have shown a high mortality rate (2% to 5%) in infants with achondroplasia and have indicated foramen magnum stenosis as the responsible factor (
33).
Radiographic Characteristics.
There are several features typically seen on the radiographs of individuals with achondroplasia, but caution should be exercised in interpreting the absence of such findings: not all affected individuals exhibit such radiographic characteristics.
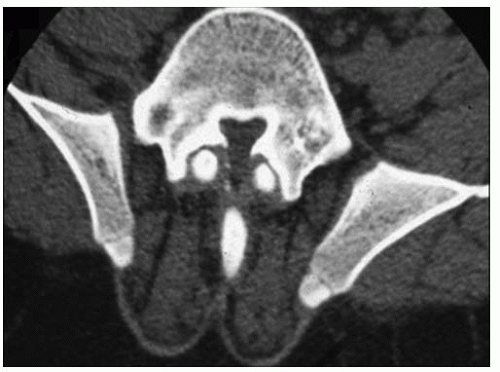
The key feature is the typical narrowing intrapedicular distance from L1 to L5 seen on the anteroposterior radiographs of affected individuals (
44,
45); in the unaffected population, the intrapedicular distance from L1 to L5 increases. The presence of such narrowing is an absolute indicator of achondroplasia, but the lack of such narrowing does not rule out the presence of achondroplasia. In addition, pedicles in those with achondroplasia are approximately 30% to 40% thicker than those in unaffected individuals (
46). In this patient population, the vertebral bodies have a scalloped appearance (
20), lumbar lordosis increases to the sacrum segment and may even become horizontal, and severe scoliosis is rare but can be seen; however, the incidence of cervical instability is not higher than that in the unaffected population (
20,
46).
Other radiographic abnormalities include underdeveloped facial bones, skull base, and foramen magnum; square iliac rings; rhizomelic shortening; and flared metaphysis of the long bones. Affected individuals also have a pronounced, inverted “v” shape of the distal femoral physis with normal distal femoral epiphysis, and the metacarpals and metatarsals are almost all equal in length. The iliac wings have a squared appearance. The metaphysis of all long bones is flared in appearance. Despite being short, the diaphyses of all long bones are thick. The sites of major muscle insertions, such as the tibial tubercle, greater trochanter, and insertion of the deltoid, are more prominent than usual. The epiphysis throughout the skeleton is normal in appearance and development; consequently, degenerative joint arthritis or changes are rarely seen.
General Medical Treatment.
Although children with achondroplasia are typically healthier than those with other dysplasias, infants and young children with achondroplasia should be closely monitored and evaluated, especially during the first few years of life, for signs and symptoms of foramen magnum stenosis (see earlier). If the diagnosis is made clinically, an MRI should be ordered to show the stenosis. At this point, neurosurgery can enlarge the foramen magnum. Sometimes, surgeons may need to perform a durotomy or an expansion of the dura and a C1 laminectomy. Many of these children also have dilatation of the veins of their cranium secondary to venous distension, which can also be relieved by such surgery. As indicated earlier, children with achondroplasia have delayed motor milestones; for example, most unaffected children walk by 12 months, whereas most of those with achondroplasia do not walk until 18 months. Postsurgery, patients typically are able to start achieving milestones much more quickly and progress rapidly (
38,
40). In addition, children with achondroplasia have a higher risk of respiratory complications than do unaffected children, not only because of midface hypoplasia and upper airway obstruction, but also because of a decreased respiratory drive that can be secondary to foramen magnum stenosis. Early brain stem decompression can decrease the risk (
37).
Otolaryngeal problems are also prevalent: 90% of the patients with achondroplasia can experience otitis media before they are 2 years old (
47), and many require ear tube placements. Adenoid and tonsil hypertrophy in the presence of midface hypoplasia can cause obstructive sleep apnea. The otitis media and adenotonsil hypertrophy may result in conductive hearing loss that can impair speech development and delay.
Achieving and maintaining an ideal body weight is also difficult and a lifelong struggle. Currently, because there are no standardized charts for size and weight, observing skin-fold thickness and noting general appearance may be the best clinical option (
48,
49).
Children with achondroplasia are typically not deficient in growth hormone levels, but there is a substantial amount of research with regard to the administration of growth hormone to supplement height (
18,
50,
51). Typically, in the first year of receiving growth hormone treatment, there is an increase in the growth height velocity, but it diminishes over the next 2 to 3 years, with a net result of no real increase. There has been speculation that too much growth hormone can hasten the development of spinal stenosis, which is one of the worst complications of achondroplasia (
18,
50,
51).
There are several other otolaryngologic problems that are usually secondary to the underdevelopment of midface skeletal structures. Maxillary hypoplasia can lead to dental overcrowding and malocclusion (
52). Many children with this condition require orthodontic attention. In such children, Eustachian tubes often do not function properly because the children are smaller than the unaffected population and more horizontally than vertically positioned, decreasing the ability to drain middle ear fluid (
53).
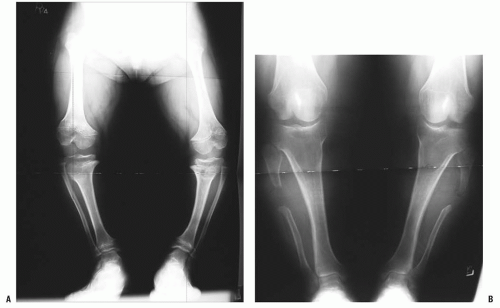
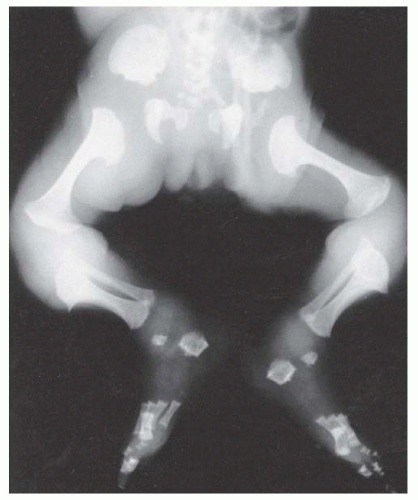
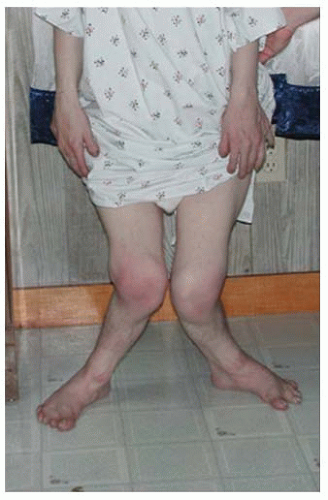
Orthopaedic problems include angular deformities of the lower extremities, genu varum at the knees, thoracolumbar kyphosis, and spinal stenosis (which can occur at any level of the spinal canal). Malalignment of the lower extremities is typically secondary to genu varum or ankle varus (
24,
54). A very small percentage of patients have genu valgum, which rarely becomes severe enough to require treatment, but genu varum may progress to cause substantial pain and difficulty in ambulation (
24,
55). Some clinicians have postulated that the longer fibula is the cause of this pain, but others have shown that the length of the fibula has no direct relationship on the amount of bowing on the knee (
56,
57 and
58) (
Fig. 7-4). Leg malalignment has been shown to be the result of ankle, distal femur, or proximal tibia deformity, or from a combination thereof. Incomplete ossification epiphysis often makes it quite challenging to elicit the source of this malalignment. Arthrograms are typically used at our institution to help identify the exact location of the deformity and are especially helpful in patients <8 years old (
12). Although bracing has been used elsewhere to help control ligamentous laxity and to try to correct bowing, we have found that the short and often pendulous nature of the legs of patients with achondroplasia makes it difficult to provide a brace with proper fit and enough of a mechanical advantage to correct the malalignment. During the past 10 years of our practice, no brace has been used to control malalignment, and surgical decisions are not made until the child is at least 3 years old. The indication for surgery is persistent pain that is secondary to malalignment (not to spinal stenosis) deformity severe enough to cause a fibular thrust, resulting in a gap between the proximal tibia and the femur on ambulation (
40,
57,
59). Again, in our practice, if the decision has been made for surgical intervention, an arthrogram is obtained to evaluate the optimal location of the osteotomy. Such arthrography also often identifies internal tibial torsion, which can then be corrected concurrently. Although fibular shortening has been advocated in the past (
55), we and others (
60,
61) do not think it is ever indicated. Treatment indications are difficult to define clearly because there are no natural history studies showing which degree of deformity causes early degeneration.
Short Stature and Limb Lengthening.
Infants with achondroplasia are shorter than other individuals and the deficit progresses until skeletal maturity.
Everyday difficulties as the result of short stature include using public restrooms, face washing in public restrooms, hair combing, engaging in hobbies involving physical activity, playing sports with average-statured individuals, conducting routine business transactions (often at countertop level), and driving a car. Nevertheless, the decision to augment stature is difficult and controversial because the procedure is time consuming, complicated (
40,
62), and fraught with complications (
38,
40).
First, surgical lengthening can achieve quite a bit of height if done safely and correctly (
13,
63,
64), but because it is a time-consuming process, it removes these children from their normal activities of school and socialization. The psychologic impact can be tremendous, especially if the lengthening goals
are not achieved. At some centers, lengthening is performed at two separate time intervals, the first typically at the age of 7 years old and the second at preadolescence. The overall time frame for surgery and postoperative therapy may be up to 3 years. Some centers prefer to delay lengthening until early adolescence to increase the patient’s participation for the rehabilitation process and also the decision making as to whether or not the lengthening should be done. We know of only one child with achondroplasia who has had limb lengthening and whose parents were also affected.
Second, surgical limb lengthening is a very complicated endeavor (
40,
62), and one that is fraught with complications (
38,
40). In one study by Aldegheri and Dall’Oca (
65), 43% of the patients who underwent limb lengthening had complications, including fracture, failure of premature consolidation, malunion, malalignment, joint stiffness, and infection. One report in the literature indicates increased symptoms of lumbar spinal stenosis (
66). The effect of limb lengthening on spinal stenosis needs additional investigation. Humeral lengthening, often is combined with lower extremity limb lengthening, may be the most functionally appreciated because it makes it is easier to perform personal care, put on shoes and socks, and perform extended reaching. The procedure also has lower risks than lower limb lengthening.
Despite the fact that limb lengthening has been a procedure in frequent use for several decades, to our knowledge, the functional benefits after elective limb lengthening have never been studied.
Spinal Aspects
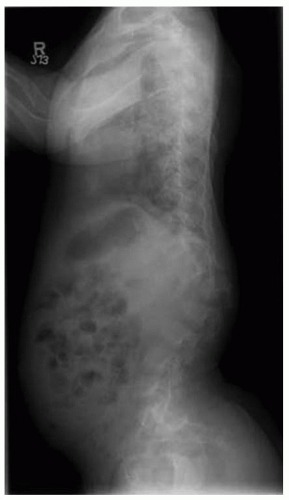
Thoracolumbar kyphosis develops in most infants with achondroplasia. A newborn with achondroplasia typically has a thoracolumbar kyphosis centered at approximately T12-L1. When sitting begins, the infant slumps forward because of trunk hypotonia, in combination with a relatively large head, flat chest, and protuberant abdomen. The apex of the vertebral deformity becomes wedge-shaped anteriorly, although it usually is a reversible phenomenon. This condition should not be confused with a diagnosis of congenital kyphosis. Most of these patients improve by the 2nd or the 3rd year of life, after walking begins and muscle strength increases (
15,
27,
67,
68). However, persistent kyphosis can increase the risk of symptomatic stenosis, putting pressure on the conus (
Fig. 7-5). To prevent persistent kyphosis, Pauli et al. (
27) recommended early parental counseling (before the infant is 1 year old) for prohibition of unsupported sitting or sitting up at more than a 45-degree angle and for the use of the following measures: firm, back-seating devices; curling the infant into a “C” position; hand counterpressure when holding the infant; and bracing as needed. In the study by Pauli et al. (
27), bracing is initiated for patients who develop kyphosis that does not correct to <30 degrees on prone lateral radiographs. Those authors initially used bracing but found it cumbersome
and not very helpful. In addition, the braced patient may be at an increased risk for falls because of the brace’s large size and the patient’s small body, poor trunk control, and developmental delay. Bracing may also have a detrimental effect on pulmonary function in children with small thoracic cages. In our practice, we have found that bracing has delayed the onset of walking. If wedging of the vertebrae persists beyond the ages of 4 or 5 years, and surgical intervention is not sought, we recommend a trial of using hyperextension casts to see if it will help with the wedging. Although some surgeons have used this technique with some benefit (
69), we have seen several instances of such use that have resulted in numerous complications, including skin breakdowns, the inability to tolerate the casts, decreased ability to ambulate, and others. Currently, the indication for safe surgical intervention in our practice is kyphosis ≥50 degrees at 5 years of age with no sign of improvement (
70). The key is twofold: (a) no hooks, wires, or any other hardware in the canal and (b) no overcorrection. Correction should limited to what is obtainable preoperatively with the awake child hyperextended laterally over a bolster. The threshold for performing an anterior arthrodesis has decreased with increased rigidity instrumentation by the placement of pedicle screws at every level (
70,
71,
72,
73,
74,
75 and
76).
For the child with achondroplasia, thoracolumbar kyphosis, and concurrent spinal stenosis symptoms, MRI will be obtained; if it shows anterior cord impingement, a corpectomy via an anterior approach will be performed. In this situation, we would not perform a vertebral body resection posteriorly because we think the achondroplastic spinal cord is not mobile enough to tolerate such a procedure. Currently, any child >3 years old who can accommodate pedicle screws, including cervical spine screws, is not placed in a cast or brace postoperatively. However, for a child <3 years old with pedicle screws, a bracing protocol is instituted for 3 months.
Lumbar stenosis typically can present during the second or third decade of life of an individual with achondroplasia, but it can be seen as early as 18 months (
Fig. 7-6). Patients typically present with complaints of lower back pain, leg pain, progressive weakness of the extremities, numbness, and tingling, symptoms that often are decreased or alleviated completely by squatting or bending over—maneuvers that reduce the lumbar lordosis, increase the size of the canal, and relieve the pressure. Surgical indications include myelopathy, progressive signs and symptoms, inability to ambulate more than one or two city
blocks without having to stop or squat to relieve the pressure. Preoperative workup includes MRI and possibly a CT scan (
Fig. 7-7). By correlating the physical examination and the MRI study, the clinician can identify the approximate level of the most severe stenosis. Usually, treatment is a wide decompression that extends at least two levels above the point of the most severe stenosis and down to the sacrum. In skeletally immature patients, a posterior spinal fusion with pedicle screw instrumentation needs to be done concurrently to prevent progressive kyphosis. If the patient is skeletally mature and has no underlying kyphosis, posterior decompression can be done alone, without a concurrent fusion (
40,
46,
71,
72,
73 and
74,
77).