Chapter 6 The Innate Immune System in SLE
Adaptive immunity and innate immunity can be distinguished from each other by the nature of the receptors involved in antigen recognition. At one end of the spectrum, we find the extreme heterogeneity of the classical T- and B-cell antigen receptors. Here, multiple gene segments are assembled through a mechanism that utilizes combinatorial diversity, junctional diversity, nontemplated base inserts, and even somatic mutation (in B cells) to generate repertoires that approximate 1013 to 1018 unique sequences and thereby allow the immune system to develop a highly tuned and sophisticated response to the world of pathogens. At the other extreme are the pattern recognition receptors (PRRs) used by dendritic cells, macrophages, neutrophils, and many other components of the innate immune system to target a broader category of molecular patterns. Importantly, lymphocytes, and especially B cells, can also express PRRs and can therefore be considered a component of the innate immune system as well as the adaptive immune system. These PRRs were originally conceived as a highly efficient surveillance system, designed to discriminate host from pathogen by detection of pathogen-associated molecular patterns (PAMPs) and thereby alert the immune system to the first signs of microbial infection.1 However, it is becoming increasingly apparent that these same PRRs can detect endogenous ligands that can be released from dead or dying cells or can be expressed on the surfaces of apoptotic cells or bodies. Although the response to such danger-associated molecular patterns (DAMPs) presumably plays a critical role in tissue repair and/or the clearance of cell debris, the failure to appropriately regulate such self-recognition can lead to serious pathologic complications. A case in point are systemic autoimmune diseases such as SLE.
What Constitutes an Autoantigen?
Autoantibodies in general target a remarkably small fraction of the general pool of mammalian proteins. This level of specificity has to be addressed by any theory that tries to explain the loss of tolerance so evident in systemic autoimmunity. Circumstantial data from a variety of studies have linked the onset and recurrence of SLE with various types of viral infections.2,3 As alluded to previously, in this context, engagement of PRRs can activate innate immune system components to produce inflammatory cytokines and chemokines and to upregulate co-stimulatory molecules. Such events could theoretically enhance the presentation of self-peptides, as well as microbial peptides, and thereby lead to a loss of tolerance. However, if such a “revved-up” immune system were the major cause of SLE, autoreactivity would be much more common and would most likely target a much broader set of self-components than we know to be the case.
The concept of molecular mimicry constitutes a second potential link between infection and autoimmune disease. Cross-reactivity between viral peptides and specific autoantigen-associated peptides has been reported by a number of groups.4 Weakly self-reactive T cells activated by the viral peptide could then further activate any antigen-presenting cells, including B cells, thereby extending the response to additional epitopes associated with a particular macromolecular complex or even to unrelated proteins located on apoptotic bodies or other aggregates of cell debris. Although attractive conceptually, this molecular mimicry model cannot explain why the same autoantibody specificities are found in populations of patients with SLE, animal models of SLE, and even germ-free autoimmune-prone mice.
A third possible pathogen-linked explanation for the break in tolerance is the creation of neoepitopes by antiviral effector mechanisms. It has been shown that many of the common autoantigens are substrates for granzymes or caspases produced by activated cytotoxic effector populations.5 The cleaved protein fragments theoretically could adopt novel conformations for which tolerance has not been established or could be processed by the antigen presentation machinery to reveal previously unavailable “cryptic” peptides. Other examples of protein modification that could result in neoepitopes are oxidation, phosphorylation, methylation, demethylation, and citrullination. Such modifications are commonly associated with tissue injury, inflammation, and various forms of cell death,6–8 conditions that can again promote immune activation.
All the preceding possibilities may well contribute to the onset of autoimmunity and may depend directly or indirectly on an activated innate immune response. However, in all cases, they imply a relatively passive role for the autoantigen per se and still fail to explain why most of the major autoantigens are recurring targets in a wide range of autoimmune conditions, including SLE. Alternatively, growing evidence accumulated over the past decade points to a much more proactive role for the autoantigen in immune activation. In fact, it is now clear that many autoantigens can either directly engage PRRs or can activate components of the innate immune system through other mechanisms and thereby act as autoadjuvants.9,10 Importantly, further identification of the relevant PRRs and their downstream signaling pathway components will point to less invasive therapeutic options than those currently available to patients.
The Endosomal Nucleic Acid–Sensing PRRs
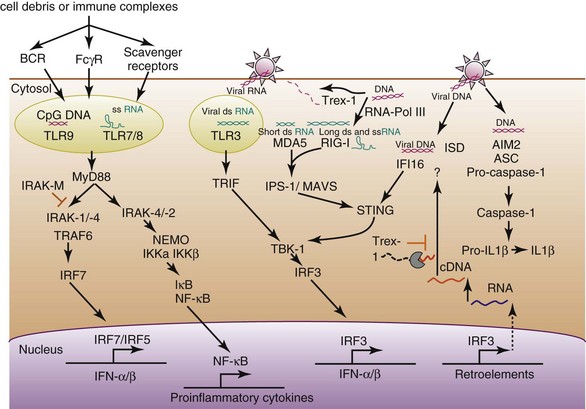
The clearest association is with members of the Toll-like receptor (TLR) family. TLRs are class I transmembrane proteins consisting of an N-terminal region made up of a tandem array of leucine-rich repeats, a transmembrane domain, and a cytosolic Toll–interleukin-1 receptor (TIR) domain responsible for downstream signaling events.11 TLRs are normally found as homodimers or heterodimers that characteristically form overlapping horseshoe-shaped ectodomains. TLRs can be divided into two categories, those that are normally expressed on the cell surface and those whose expression is predominantly limited to intracellular compartments—endoplasmic reticulum (ER), endosomes, and lysosomes. The endosomal receptors, TLR3, TLR7, TLR8, and TLR9, recognize either RNA or DNA, and are the TLRs primarily involved in viral immunity. In order for these endosomal receptors to traffic from the ER to the endosomal/lysosomal compartments, they need to associate with the chaperone protein Unc93b. Murine or human cells that express a nonfunctional form of Unc93b cannot respond to any of the ligands normally detected by the endosomal receptors.12,13 Cathepsins active in low-pH compartments cleave TLR9 and TLR7, and this cleavage is thought to enhance ligand recognition.14,15
Because these TLRs are located in intracellular compartments, and not on the cell surface, one major factor that limits their activity is ligand accessibility—nucleic acids need to colocalize with the TLRs in the appropriate endosomal/lysosomal compartment in order to engage these receptors. Autoantigen trafficking to these compartments is mediated by cell type–specific cell surface receptors. Dendritic cells and neutrophils depend on Fc-gamma receptors (FcγR) to bind autoantigen/autoantibody immune complexes and then transport the complexes to endosomes. In B cells, this transport role is facilitated by the B-cell receptor (BCR).16 Nucleic acid–associated cell debris, perhaps in the form of apoptotic bodies or microvesicles, can also be taken up by phagocytic cells via various scavenger receptors. In addition, delivery of nucleic acids to the right compartment can be facilitated by antimicrobial peptides such as LL37.17
TLR9 is the main sensor of DNA and was originally thought to distinguish microbial DNA from mammalian DNA, on the basis of reactivity with so-called hypomethylated CpG motifs, which are rarely found in mammalian DNA but are common in bacterial and viral DNA. Later studies have clearly demonstrated that under the appropriate circumstances, TLR9 can also detect mammalian DNA. Nevertheless, DNA sequence is still relevant because mammalian DNA sequences enriched for CpG dinucleotides are more potent activators of TLR9 than DNA sequences devoid of CpG dinucleotides.18 Potential sources of immunostimulatory mammalian DNA include CpG islands, mitochondrial DNA, and retroelements.
TLR7, TLR8, and TLR3 are the RNA-sensing TLRs. TLR7 and TLR8 were initially identified by their ability to respond to synthetic antiviral compounds such as imidazoquinoline derivatives and guanine analogs with strong type I IFN–inducing activity. TLR3 was identified by its capacity to bind a synthetic analog of double-stranded RNA, polyinosinic-polycytidylic acid (poly(I:C)), another mimic of viral infection and a strong inducer of IFN. These receptors bind various forms of single-stranded (ss) or double-stranded (ds) viral RNAs, respectively, and failure to express functional forms of each of these RNA-sensing receptors is associated with susceptibility to very specific types of viral infections. As in the case of TLR9, the RNA-reactive TLRs can also detect mammalian RNAs.19,20 Here again, sequence and structure are most likely key determinants of ligand avidity, because TLR7 and TLR8 preferentially bind unmodified uridine (U)–rich ssRNAs. Many of the small RNAs associated with the macromolecular structures that include common autoantibody targets fit this category.
Both TLR9 and TLR7 are constitutively expressed by plasmacytoid dendritic cells (pDCs). Although a relatively rare DC population, pDCs can produce extremely high levels of IFN-α in response to both exogenous (viral) and endogenous inducers. Importantly, TLR ligands also induce pDCs to make proinflammatory cytokines such as tumor necrosis factor alpha (TNF-α) and interleukin (IL) 6. B cells also express TLR9 and TLR7, although B-cell expression of TLR7 is markedly increased by type I IFNs. The role of TLR8 in mice is controversial, with data to suggest that it is relatively nonfunctional or a negative regulator of other TLRs. In humans, TLR8 is found predominantly on myeloid-derived cells, where it can also lead to the production of inflammatory cytokines. TLR7, TLR8, and TLR9 signaling pathways depend on the adaptor protein MyD88, on downstream components IRAK4, IRAK1, and IRAK2, and on TRAF6 (TNF-receptor associated factor 6) to activate interferon regulatory factors IRF7 and IRF5 as well as the nuclear factor kappa B (NF-κB) pathway to promote both the production of IFN and proinflammatory cytokines, respectively. TLR3 is expressed by both hematopoietic and nonhematopoietic cells (fibroblasts) and works through the adaptor protein TRIF to activate the transcription factors IRF3 and NF-κB, also leading to the production of IFN and proinflammatory cytokines (Figure 6-1).






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


