Chapter 7 The Environment in the Pathogenesis of Systemic Lupus Erythematosus
INTRODUCTION
Human lupus is a chronic relapsing autoimmune disease characterized by autoantibodies to a host of cellular components and immune complex deposition in the kidney and other tissues. Although the etiology of lupus is unknown, both genetic and environmental factors are implicated in disease pathogenesis. Evidence for a genetic contribution comes from familial aggregation of autoimmunity in approximately 20% of lupus cases,1 a higher concordance rate in monozygotic (∼25%) relative to dizygotic twins (2%),2 the evidence for linkage at multiple loci across the human genome as shown by genome-wide scans of subjects with familial lupus,3 and the discovery of various susceptibility genes for the disease.4 The observations that the majority of lupus is sporadic and that drugs such as procainamide, hydralazine, and others (as well as UV light) trigger lupus-like autoimmunity, and the lack of complete concordance in identical twins, indicate a prominent role for exogenous agents.5 How environmental agents interact with the various genetic loci to produce autoantibodies, the hallmark of lupus, is incompletely understood. Current models postulate that autoimmunity develops in genetically susceptible hosts exposed to appropriate environmental triggering factors. However, the nature of the interactions among various genetic elements and the environment is yet to be revealed. Herein, we discuss selected environmental factors associated with systemic lupus erythematosus (SLE), and review the evidence for their pathogenic role in the disease. The environmental factors discussed include drugs, UV light exposure, infectious agents, chemicals, heavy metals, and dietary factors.
DRUGS
Drug-induced lupus provides the clearest example of an exogenous agent causing lupus-like autoimmunity. In the majority of cases, withholding the offending medication results in the disappearance of autoantibodies and resolution of disease signs and symptoms, and cautious readministration causes disease recurrence,6 supporting cause and effect. More than 100 drugs have been implicated in causing lupus and lupus-like syndromes. Whereas some of the reports are anecdotal, others (such as hydralazine and procainamide) are well established by clinical studies.5 A recent controlled case study utilizing the General Practice Research Database in the United Kingdom has now estimated the relative risk for developing SLE in 1437 patients exposed to 13 of the known lupus-inducing drugs and 6963 matched controls. This study found supportive results for the majority,7 providing additional epidemiological evidence for an association between specific drugs and lupus-like diseases (see Table 7.1).
TABLE 7.1 DRUGS MOST COMMONLY IMPLICATED IN DRUG-INDUCED LUPUSa
| Medication odds ratio | 95% Confidence interval | |
|---|---|---|
| Acebutolol | 8.2 | 1.4–46.7 |
| Captopril | 2.4 | 1.5–4.1 |
| Carbamazepine | 3.4 | 2.3–4.8 |
| Chlorpromazine | 2.7 | 1.4–5.2 |
| Hydralazine | 6.7 | 1.9–23.6 |
| Isoniazide | 20 | 2.2–178.9 |
| Methyldopa | NA | |
| Minocycline | 4.3 | 3.1–6.0 |
| Penicillamine | 29.6 | 6.6–132.1 |
| Procainamide | NA | |
| Propylthiouracil | NA | |
| Quinidine | 5.0 | 0.7–35.5 |
| Sulfasalazine | 39.9 | 17.1–93.2 |
NA: Not available or the study was underpowered to detect any difference.
a. Odds ratio for developing lupus upon exposure is indicated where available.7
Because the inciting agent is known, drug-induced lupus provides a unique opportunity to identify disease mechanisms potentially relevant to idiopathic lupus. Mechanistic studies have often focused on hydralazine and procainamide, recognized as causing lupus-like autoimmunity since 1953 and 1962, respectively.8 Recent studies demonstrate that these drugs modify epigenetic mechanisms regulating T lymphocyte gene expression, with important implications for the pathogenesis of both drug-induced and idiopathic lupus. Common to both forms of lupus is dysregulation of DNA methylation, an epigenetic mechanism regulating gene expression.
DNA Methylation in Drug-Induced Lupus
DNA Methylation
DNA methylation in vertebrates refers to the post-synthetic methylation of cytosine bases in DNA to form methylcytosine. Methylcytosine is usually found in CG pairs, and most CG pairs in the mammalian genome are methylated. The exceptions are those in promoter sequences of transcriptionally active genes. Promoters of most active genes are hypomethylated, and methylation of the promoter sequences renders the genes transcriptionally inactive.9 DNA methylation suppresses gene expression by several mechanisms. These include inhibiting binding of some transcription factors10,11 and attracting methylcytosine binding proteins that sterically inhibit transcription factor binding, as well as tether chromatin inactivation complexes to methylated sequences. The chromatin inactivation complexes promote localized chromatin condensation into an inactive configuration,12 and may be the most transcriptionally important mechanism.
Methylation patterns are established during differentiation and serve to suppress genes unnecessary or detrimental to the function of each specific cell type. DNA methylation is also involved in female X chromosome inactivation, genomic imprinting, and suppressing parasitic DNA. The importance of DNA methylation is evidenced by studies demonstrating that homozygous deficiency of any of the methyltransferases results in death during embryogenesis or in the early postnatal period, and that changes in established methylation patterns causes changes in gene expression that contribute to aging and cancer.13
De novo methylation of previously unmethylated DNA, such as occurs during differentiation, is mediated by DNA methyltransferases Dnmt3a and Dnmt3b. Methylation patterns are then replicated during mitosis by the maintenance methyltransferase Dnmt1. Dnmt1 binds proliferating cell nuclear antigen (PCNA) at the replication fork, recognizing and methylating hemimethylated DNA that results from a methylated parent strand and unmethylated daughter strand, while ignoring unmethylated DNA, thereby replicating methylation patterns.13 Dnmt1 expression is linked to the cell cycle, and is regulated in part via signaling through the extracellular signal-regulated kinase (ERK) and Jun N-terminal kinase (JNK) pathways.14,15
Inducing DNA Demethylation
DNA can be demethylated using DNA methyltransferase inhibitors such as 5-azacytidine. 5-azacytidine is a cytosine analogue that is incorporated into DNA during S phase, where it covalently binds DNA methyltransferases during maintenance DNA methylation.16 This depletes cellular DNA methyltransferases with subsequent genome-wide hypomethylation of the newly synthesized DNA and activation of those genes repressed by methylation and for which the cell expresses the necessary transcription factors.
Effects of 5-azacytidine on T-cell Function and Gene Expression
5-azacytidine has been used to characterize how DNA methylation regulates T-cell function. Treating human and murine cloned and polyclonal antigen-specific CD4+ T-cells with 5-azacytidine causes loss of the requirement for nominal antigen and the ability to respond to self class II MHC determinants without added antigen. The autoreactive cells also acquire the ability to promiscuously kill autologous or syngeneic macrophages and overstimulate B-cell antibody production.17 This autoreactive response resembles the response of semiallogeneic CD4 T-cells to host class II determinants in chronic graft-versus-host disease, which causes an autoimmune disease with features of lupus in mice.18 The similarities between 5-azacytidine-induced CD4+ T-cell autoreactivity and CD4+ alloreactive responses to class II MHC molecules is further supported by studies demonstrating that 5-azacytidine-treated CD4+ T-cells cause a lupus-like disease in syngeneic recipients. The mice develop autoantibodies to single- and double-stranded DNA, a lupus band test and nephritis, resembling chronic graft-versus-host disease.19
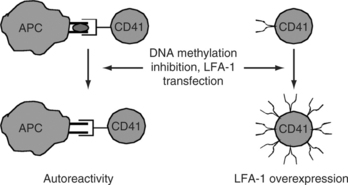
T-cell genes affected have been identified with mechanism-focused approaches and more recently via oligonucleotide arrays. Genes modified by 5-azacytidine and relevant to autoimmunity include CD11a (LFA-1a), perforin, CD70, and INF-γ,17,20 among others. CD11a is a subunit of LFA-1 (CD11a/CD18), an adhesion molecule expressed on most hematopoetic cells. 5-azacytidine demethylates a series of alu repeats 5′ to the CD11a promoter, increasing transcription.21 CD11a demethylation participates in the autoreactivity of the treated cells through increased LFA-1 expression. Increasing LFA-1 expression by transfection of human or murine CD4+ T-cells causes the same loss of restriction to nominal antigen and responses to self class II MHC molecules without specific antigen (as seen following treatment with DNA methylation inhibitors).21 The autoreactivity may be due in part to overstabilization of the low-affinity interaction between the T-cell receptor and self class II MHC determinants without specific antigen, thereby decreasing the threshold for activation.23 Murine CD4+ T-cells made autoreactive by LFA-1 transfection cause a lupus-like disease in vivo resembling the autoimmunity caused by injecting demethylated LFA-1 overexpressing cells,24 further supporting the importance of autoreactivity caused by this mechanism (see Fig. 7.1).
Demethylated autoreactive CD4+ T-cells also acquire the ability to kill autologous or syngeneic macrophages. Aberrant perforin expression contributes to this autoreactive cytotoxic response. Perforin is a cytotoxic molecule normally expressed by NK cells and cytotoxic CD8+ T-cells. Demethylating a conserved region linking the perforin promoter to an upstream enhancer induces perforin expression in CD4+ T-cells and increases perforin expression in CD8+ T-cells.25 Concanamycin, a perforin inhibitor, decreases autoreactive macrophage killing by hypomethylated CD4+ cells, suggesting that perforin contributes to this cytotoxic response.26 The macrophage killing may contribute to autoantibody formation in the demethylation model by depleting scavenger cells responsible for clearing apoptotic debris while simultaneously increasing the release of apoptotic material, both of which cause lupus-like autoimmunity in animal models.27,28
CD70 and IFN-γ overexpression in hypomethylated CD4+ T-cells may contribute to autoimmunity by increasing antibody production. CD70 is expressed on stimulated T-cells, and interacts with CD27 on B-cells to promote immunoglobulin synthesis and secretion. 5-azacytidine increases CD70 by demethylating a region just upstream of the active promoter.29 CD70 overexpression on CD4+ T-cells treated with 5-azacytidine causes B-cell overstimulation and increased IgG secretion30 resembling the polyclonal B-cell activation seen in the peripheral blood B-cells of patients with lupus.31 In addition, IFN-γ is suppressed in Th2 cells by promoter methylation and is demethylated with 5-azacytidine, inducing expression.32 IFN-γ similarly stimulates B-cell immunoglobulin secretion, and lupus B-cells secrete significantly greater amounts of IgG in response to IFN-γ than controls.33 The effects of DNA hypomethylation on T lymphocyte function and gene expression are summarized in Fig. 7.2.
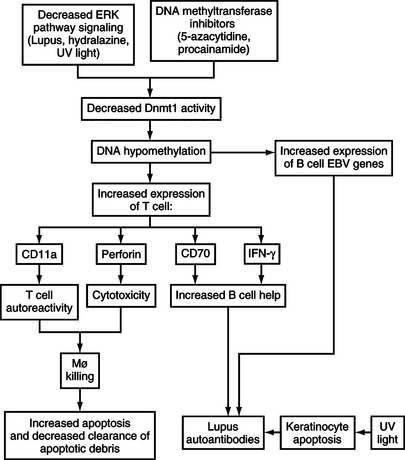
Finally, 5-azacytidine can reactivate endogenous retrovirus expression in T-cells34 and Epstein-Barr virus proteins as well as the Epstein-Barr viral lytic cycle in B-cells.35 EBV and endogenous retroviral genes are normally suppressed by DNA methylation, and their reactivation has been associated with the development of lupus-like autoimmunity.34,36 The possible role of EBV and retrovirus expression in lupus is discussed further in material following and elsewhere in this textbook.
DNA Demethylation by Lupus-Inducing Drugs
The observation that inhibiting DNA methylation causes lupus-like autoimmunity suggests that drugs causing lupus-like autoimmunity might inhibit DNA methylation. Indeed, when CD4+ T-cells are treated during S phase with either hydralazine or procainamide the cells become autoreactive, similar to what is seen with 5-azacytidine.37 Low concentrations (10−7 M) of hydralazine and procainamide induce autoreactivity, which increases in a dose-dependent fashion. Interestingly, the concentrations of hydralazine and procainamide causing autoreactivity fall in the therapeutic concentrations of these drugs and are similar to the concentrations that induce lupus. Hydralazine and procainamide also increase LFA-1 and CD70 expression, similar to 5-azacytidine.30,38
Consistent with the LFA-1 overexpression and autoreactivity, hydralazine and procainamide also inhibit methylation of newly synthesized DNA in human T-cells.37 Procainamide is a competitive T-cell DNA methyltransferase inhibitor.39 In contrast, hydralazine inhibits the extracellular signal-regulated kinase (ERK) pathway (thereby decreasing DNA methyltransferase expression) but does not directly inhibit DNA methyltransferase enzyme activity.40 DNA methyltransferase 1 and 3a levels are also decreased in murine T-cells treated with the ERK pathway inhibitor U0126, and the treated cells have hypomethylated DNA (similar to human T-cells treated with hydralazine).40 To determine if inhibiting the ERK pathway in T-cells causes autoreactivity, D10 cells, (cloned conalbumin reactive Th2 line from AKR mice) were treated with the MEK inhibitor U1026 or hydralazine. U1026- and hydralazine-treated cells overexpressed LFA-1 and responded to syngeneic antigen-presenting cells in the absence of the antigen, thereby resembling T-cells treated with 5-azacytidine (Fig. 7.1).
In vivo effects of cells made autoreactive with procainamide and hydralazine have been similarly determined in murine models. When polyclonal CD4+ T-cells from DBA/2 mice are treated with 5-azacytidine or procainamide and then injected into unirradiated syngeneic female mice, the mice develop anti-DNA and antihistone antibodies, a positive lupus band test, and an immune complex glomerulonephritis.19 This experiment has been repeated using D10 cells treated with 5-azacytidine. The treated cells overexpress LFA-1, become autoreactive, and produce large amounts of IL-6.41 Adoptive transfer of the treated cells into female AKR mice induces autoantibodies directed against single-stranded and double-stranded DNA as well as antihistone antibodies. Histologic examination reveals that these mice develop immune complex glomerulonephritis, pulmonary alveolitis, central nervous system pathologies (including fibrinoid necrosis, karyorrhexis, and meningitis), and bile duct proliferation with periportal inflammation similar to primary biliary cirrhosis.41 The same model has been used to demonstrate that procainamide is more potent than N-acetylprocainamide in inducing LFA-1 overexpression and autoreactivity in vitro and autoimmunity in vivo, and that hydralazine is more potent than phthalazine (the parent compound) in the same assays.38 D10 cells treated with the ERK pathway inhibitor U0126 or hydralazine and then adoptively transferred into nonirradiated syngeneic female mice, also induce the production of anti-double-stranded DNA antibodies, confirming that ERK pathway inhibition can induce autoimmunity.40
Together, these studies indicate that T-cells demethylated by treatment with either hydralazine or procainamide are sufficient to induce a lupus-like disease in vivo. This suggests a mechanism that might contribute to the development of some forms of drug-induced lupus.
DNA Methylation in Idiopathic Lupus: What We Learned from Drug-Induced Lupus
The studies on DNA methylation and drug-induced lupus prompted confirming studies in patients with idiopathic SLE. The results suggest that idiopathic lupus may be caused by mechanisms very similar to those by which procainamide and hydralazine cause lupus-like autoimmunity. Early studies demonstrated that T-cells from lupus patients have a genome-wide reduction in methylated DNA and decreased DNA methyltransferase enzymatic activity.42 Subsequent studies revealed that lupus T-cells have decreased levels of Dnmt1 transcripts, due to defective ERK pathway signaling, similar to hydralazine-treated T-cells.14 Examination of the previously identified methylation-sensitive genes revealed that CD4+ lupus T-cells aberrantly overexpress CD11a, CD70, and perforin (similar to experimentally demethylated T-cells),26,30,43 that LFA-1 and perforin overexpression contribute to the spontaneous macrophage killing characteristic of lupus T-cells,26 and that CD70 overexpression contributes to overstimulation of autologous B-cell IgG production.30 Moreover, the same CD11a, CD70, and perforin promoter sequences demethylated by methylation inhibitors are demethylated in lupus T-cells.21,25,29
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


