Chapter 6 Overview of the Pathogenesis of Systemic Lupus Erythematosus
INTRODUCTION
The current models of inheritance of disease susceptibility of multifactorial traits such as SLE favor the principle of “threshold liability.“1,2 According to this model, the genetic makeup of an individual who is predisposed to developing SLE comprises a certain number of SLE susceptibility genes that contribute to additive disease liability when their number exceeds a certain hypothetical threshold. This “additive inheritance” is possibly modified further by “multiplicative inheritance,” such as epistatic interactions among susceptibility alleles, and together they skew an individual’s disease liability toward a critical threshold at which point the disease manifests.1,2 Environmental and stochastic events experienced by an individual in his life and hormonal factors could also contribute to these factors.
The pathogenesis of SLE is equally complex involving multiple immune abnormalities including abnormal B- and T-cell function that perpetuates autoantibody production by B cells and generates autoreactive T cells. In addition, abnormal clearance of immune complexes that results in their deposition in tissues, activation of complement and defective cellular apoptosis that generates a pool of potential autoantigens, are also integral components of the SLE pathology. The net result of these processes is induction of varying degrees of organ inflammation and failure, most importantly of the kidneys, heart, skin and nervous system that ultimately result in various degrees of morbidity and mortality.3 In this section, we briefly consider the pathogenesis of SLE at the molecular level. The reader is referred to appropriate chapters where each topic is discussed in detail.
ETIOPATHOGENESIS OF SLE
Environmental Factors
Environmental factors may be involved in triggering the onset of the autoimmune process in SLE in a genetically predisposed individual. These factors include drugs, UV rays in sunlight, heavy metals and chemicals, pathogenic organisms, and lifestyle, including diet. The role of these agents is considered in detail in Chapter 7.
Several drugs have been implicated in drug-induced lupus, with procainamide and hydralazine being the best studied. These drugs modify epigenetic mechanisms that control gene expression in T cells such as inhibition of DNA methylation.4 DNA methylation is a mechanism employed by cells to regulate transcription of genes, and hypomethylation of DNA could result in abnormal expression of genes implicated in the pathogenesis of SLE. The molecules that become overexpressed in helper cells include LFA-1, CD70, and IFN-γ.4 As a result, there is loss of major histocompatibility complex (MHC) restriction to self-antigens by T cells, abnormal TCR signaling, and alteration in B-cell responses that include increased antibody production.
Another environmental factor that has been known to trigger SLE flares or augment the pathologic process is UV ray exposure. Although the precise mechanism of action of UV rays is unclear, there is emerging evidence that similar to drugs that induce lupus, UV rays might also be involved in altering DNA methylation, and thus similarly alter the immune response. In addition, they might also play a role in increasing apoptosis in SLE patients and trigger the autoimmune process by unmasking potential autoantigens (reviewed in Mok and Lau5).
Among numerous chemicals and heavy metals implicated in SLE pathogenesis, the most important are crystalline silica and mercury. Their precise role in initiating/augmenting the abnormal autoimmune response remains unclear (see Chapter 7).
It is a common clinical observation that production of autoantbodies and subsequent development of SLE occurs following an infection. Several mechanisms are believed to be involved in virus-induced autoimmunity such as molecular mimicry (production of antigenic epitopes similar to self-antigens), alteration of the immune response, and other mechanisms. These mechanisms are discussed in detail in Chapters 2 and 11. Although several viruses have been implicated, the association of lupus with Epstein-Barr virus (EBV) is the most extensively studied. The characteristics of EBV that might possibly contribute to SLE pathogenesis include (1) establishment of a life-long infectious process in the host that is punctuated by periods of reactivation, (2) production of viral proteins similar to host molecules such as IL-10, CD40, and bcl-2 that have the potential to alter the immune response, (3) molecular mimicry that results in targeting of host proteins by autoantibodies, and (4) its effect on activation and immortalization of B cells.6 Cytomegalovirus (CMV), similar to EBV, is another herpes virus believed to be involved in the pathogenesis of SLE. CMV is believed to alter the immune process by multiple mechanisms such as (1) production of a protein similar to IL-10, (2) induction of cytokines such as TNF-α in massive amounts or (3) by triggering the generation of autoantibodies, the most significant among them being against the U1 small nuclear ribonucleoprotein.6 Other viruses implicated are retroviruses that share several features of herpes viruses, and parvovirus B19. However, further studies are necessary to fully ascertain their association with disease development in SLE.
The role of diet and SLE remains unresolved. Flare-up of lupus activity following ingestion of alfalfa has been reported in humans. Alfalfa contains L-canavanine that is implicated in mediating this effect, although these findings have not found sufficient support among studies in various population groups (reviewed in Mok and Lau5).
Genetic Factors
The observations that SLE aggregates in families and demonstrates a high sibling recurrence risk ratio and high disease concordance rates in identical twins, strongly support a genetic basis of susceptibility to SLE. Over the past several years, association studies of candidate genes and linkage analysis have identified several genes with varying abilities to confer susceptibility to SLE (see Chapter 8 for full discussion).
The genes coding for MHC have been associated with SLE for several decades. It has been reported that the HLA-DR2 and HLA-DR3 have been associated with a two- to three-fold relative risk conferred by each allele in the white population.7 While the mechanisms that are responsible for the contribution of these alleles to lupus pathogenesis remain unclear, the most plausible explanation is altered antigen presentation to CD4 T cells.
Another set of alleles with strong association with SLE is the Fcγ receptor genes. Allelic variants of Fcγ receptor genes can contribute to SLE pathology by altering the functions of phagocytic cells via altered binding affinity to respective subclasses of IgG. Single-nucleotide-gene polymorphisms (SNP) of FcγRIIA, FcγRIIIA, FcγRIIIB, and FcγRIIB, a cluster of four genes at 1q23 encoding for low-affinity IgG receptors, have been found to be associated with SLE, with FcγRIIA and FcγRIIIA bearing the strongest association.8–10
Complement gene products participate in rapid clearance of apoptotic debris, thus masking potential autoantigens. Complete deficiency of complement components C1q, C4, and C2 tremendously increases the risk of developing SLE in an individual. In addition, deficiencies of C1r/s, C5, and C8 have also been reported to induce SLE-like syndromes.9
Cytotoxic T-lymphocyte antigen (CTLA-4) normally serves to dampen the immune response by acting as a negative regulator of T lymphocytes. A strong association of CTLA-4 gene polymorphisms with susceptibility to SLE has been reported. Specifically an allelic variation characterized by T/C substitution at the −1722 site has been shown to influence susceptibility to SLE (reviewed in Nath et al.9 and Croker and Kimberly10).
Programmed cell death-1 (PDCD-1) is an immunoreceptor of the CD28 family normally expressed on the surface of activated T and B cells and regulates peripheral tolerance. In the European and Mexican populations, development of SLE has been attributed to a SNP in the intronic sequences of PDCD-1. This polymorphism has been shown to alter the binding site for the runt-related transcription factor 1 (RUNX1), and thus disrupt the regulation of expression of PDCD-1 protein. This process then triggers increased responsiveness of lymphocytes in SLE (reviewed in Nath et al.,9 Croker and Kimberly,10 and Shen and Tso11).
SNP have been reported for several other genes in SLE that associate these genes with SLE. Among them are protein tyrosine phosphatase N22 (PTPN22), C-reactive protein (CRP), mannose-binding lectin (MBL), cytokine genes such as tumor necrosis factor (TNF), and interferon-α (IFN-α) genes that may function by disrupting either the innate or adoptive arm of the immune response (reviewed in Nath et al.,9 Croker and Kimberly,10 and Shen and Tso11).
While significant differences exist between human and murine lupus, animal models have served as valuable tools to evaluate the genetic basis of pathology of human SLE. The murine models of SLE such as the (NZB × NZW)F1, BXSB, and MRL mice are a few such examples. Chapters 17 and 18 categorize these mouse models into groups and discuss each model and the insights we have gained from them. In a nutshell, these models have helped in narrowing down the search for candidate genes to specific lupus susceptibility loci. It is expected that with the availability of powerful genetic and proteomic tools, the animal models will aid identification of disease susceptibility genes through positional cloning and in vivo complementation studies using bacterial artificial chromosome (BAC) transgenic technology.
Role of Hormones
Given the strong gender bias observed in SLE, the role of hormonal influences in the pathogenesis of SLE has been long suspected. In general, it has been shown that androgens are immunoprotective whereas in a number of autoimmune diseases, estrogens are involved either as immunoprotective agent (especially the diseases demonstrating a Th2-type response) or as an agent involved in the disruption of tolerance. Estrogens act via ER-α and -β receptors that are expressed singly or in combination in cells of the immune system. Both T and B cells have been shown to express ER-α and -β.12 The precise mechanism by which estrogen exerts its role in SLE pathogenesis is unclear. Indeed, treatment of SLE T cells but not normal T cells results in increased activity of calcineurin phosphatase induction and expression of CD154. It has been shown that estrogen can bind to DNA directly to alter transcription of several genes and in addition may also indirectly modulate transcription via its association with other activators or repressors or through stimulation of the Erk (MAP kinase) pathway.12 Estrogen targets several genes including those that code for cytokines and molecules involved in the apoptotic factors.12 Extensive research on animal models and trials with receptor antagonists are expected to shed light on the precise role of estrogen in SLE pathogenesis.
The role of other hormones is even less clear. Important among them is prolactin, which stimulates disease activity and enhances T-cell proliferation and B-cell maturation. Estrogen may also control prolactin, thereby aiding these events indirectly. These and other hormones are discussed in detail in Chapter 9.
IMMUNOPATHOLOGY
Lymphocyte Abnormalities
T-Cell Abnormalities
Several aspects of the abnormal regulation of T cells contribute to autoimmunity in SLE. These include disruption of immune tolerance, abnormal response to autoantigens, abnormal display of autoantigens, and pathologic alterations in signal transduction across the T-cell receptor (TCR). In contrast to T cells derived from a healthy individual, SLE T cells display an activated phenotype characterized by surface expression of activation markers, lowered threshold of activation, and altered co-stimulation requirements. The activated phenotype of T cells in lupus has been reported in both humans and mice.13,14 (See Chapter 10.)
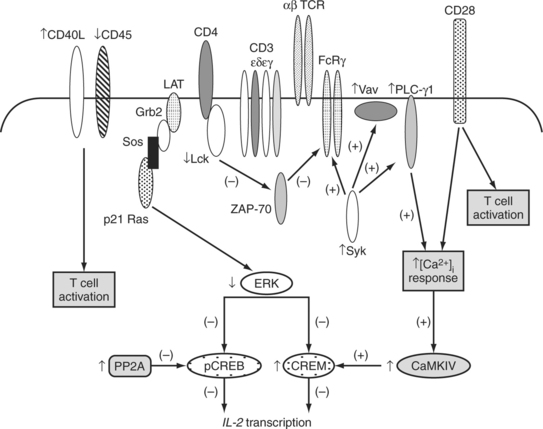
Studies that examined the structure and associations of the TCR/CD3 complex in SLE have shed light on some of the mechanisms behind the hyperexcitable phenotype of SLE T cells. These findings are summarized inFig. 6.1. Specifically, in SLE T cells, the classical TCRs that contain TCR ζ chains are replaced by TCRs that associate with the TCR ζ homologue FcRγ chain, which becomes up-regulated in SLE.15 Heightened amplification of signals emerging from this “rewired” TCR/CD3 complexes containing FcRγ is mediated via the association of FcRγ with Syk that is more potent enzymatically compared to ZAP-70 kinase, which traditionally associates with TCRs containing TCR ζ chains.13
< div class='tao-gold-member'>

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


