Chapter 17 What Do Mouse Models Teach Us about Human Systemic Lupus Erythematosus?
INTRODUCTION
Systemic lupus erythematosus (SLE) is an autoimmune disease of complex etiology primarily characterized by the presence of high titers of autoantibodies directed to many nuclear as well as cytoplasmic antigens and end-organ damage. Antibody and immuno-complex—mediated inflammation in this disease can lead to the development of glomerulonephritis, dermatitis, serositis, and vasculitis. It is estimated to affect about 1 in 2000 people with a strong gender bias (female to male ratio is about 9:1). Furthermore, blacks and Hispanics are approximately two to four times more likely to develop this disease than whites. Both genetic and environmental factors contribute to its pathogenesis.1–4 Although deficiencies in some of the early components of the complement cascade, such as C1 and C4,2,5 can lead to the development of SLE, this disease is typically polygenic in origin.4
SPONTANEOUS MURINE LUPUS MODELS REPRODUCE COMPLEXITY OF HUMAN SLE
Spontaneous murine lupus models were the earliest to be reported (Table 17.1). Several inbred and hybrid strains were noted to develop elevated levels of antinuclear antibodies (ANA) with varying degree of lupus-like kidney disease. Studies performed with these models have provided important insights into lupus pathogenesis. Some of the best-characterized models include the hybrids of New Zealand black (NZB) and New Zealand white (NZW) mice, as well as the closely related recombinant inbred NZM2410 strains.6–8 Numerous susceptibility loci have been mapped in these strains.4,9–14 The MRL/lpr and BXSB/Yaa strains also develop lupus.15–17 MRL/lpr mice carry the lpr mutation of Fas on the lupus-prone MRL background, while BXSB/Yaa mice bear the Y-linked autoimmune accelerator (Yaa) gene on the lupus-prone BXSB background. Besides these two loci, several additional loci have recently been identified in their background genomes.18–21 Likewise, (SWR × NZB)F1 (or SNF1) mice develop lupus nephritis that is clinically very similar (in onset, severity, female bias, and pathology) to the BWF1 model, as a consequence of multiple SWR and NZB loci.22–26 In these models, although disease susceptibility loci appear to be scattered randomly over all 19 autosomes, loci on four chromosomes appear to have been repeatedly mapped in several independent studies: chromosomes 1, 4, 7, and 17. Among these loci, the syntenic counterparts of the murine loci on chromosome 1 (Sle1, Nba2, etc.) and chromosome 17 (Sle4, Lbw1, etc.) have also been implicated in linkage analysis to be associated with human SLE.3,4 However, it is presently unknown if the same genes within these intervals are responsible for both the murine and human disease.
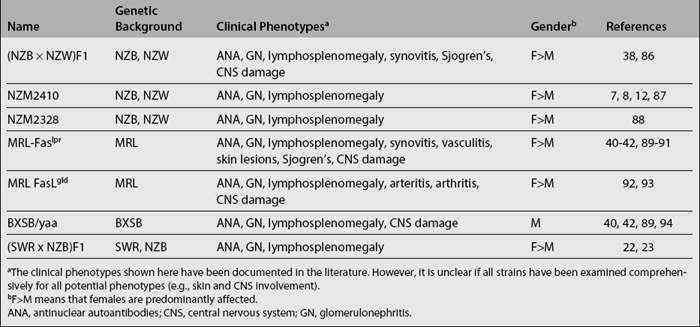
It is also interesting to consider how well these mouse models of spontaneous lupus mimic the human disease. All of the mouse models exhibit high titers of IgG anti-dsDNA and antiglomerular antibodies, accompanied by severe glomerulonephritis, resembling human SLE. Beyond this commonality, the various models reflect certain aspects of the human disease fairly well. Thus, whereas the BWF1 and SNF1 models exhibit the female bias in disease prevalence (just as in human SLE), many of the other strains do not. In fact, the BXSB strain is rather unusual in afflicting males predominantly. Whereas the MRL/lpr and BXSB models are heavily dependent on single-gene accelerators, the other models (and human SLE) do not appear to be so. Likewise, the massive degree of lymphoproliferation that characterizes the former models is also not a typical feature of lupus in other models or in humans. On the other hand, the joint and skin diseases exhibited by MRL/lpr mice represent features seen in human SLE but rarely noted in the other mouse models.27–30 More recently, it has become apparent that these mouse models may also be suitable for studying CNS lupus, a common feature of human SLE.31–43
CONGENIC MURINE LUPUS MODELS PROVIDE UNIQUE TOOLS FOR DISSECTING LUPUS PATHOGENESIS
Although linkage analyses have identified numerous loci that are associated with various disease phenotypes, they have not provided any mechanistic insights into how these loci actually contribute to these phenotypes. Congenic dissection is a strategy in which individual loci that contribute to a polygenic disease (such as lupus) can be segregated into a collection of unique substrains, each bearing an individual locus; one is thus allowed to study the component phenotypes contributed by each locus separately. Using this strategy, various lupus susceptibility loci have been successfully introgressed onto the genome of lupus-resistant strains, such as the C57BL/6 (B6) (Table 17.2). These newly generated congenic mouse strains constitute a unique and powerful tool for dissecting lupus pathogenesis. Since most lupus congenics derived to date have been generated on the B6 background, one can easily breed them to existing mouse tools such as transgenics and knockouts (involving various molecules of immunologic importance), many of which are already on the B6 background. This would then allow the scientist to study the roles of specific cells or molecules in the context of different lupus susceptibility loci. Moreover, the same breeding approach used to create congenic strains can be repeatedly applied in order to further “narrow” the relevant congenic intervals.
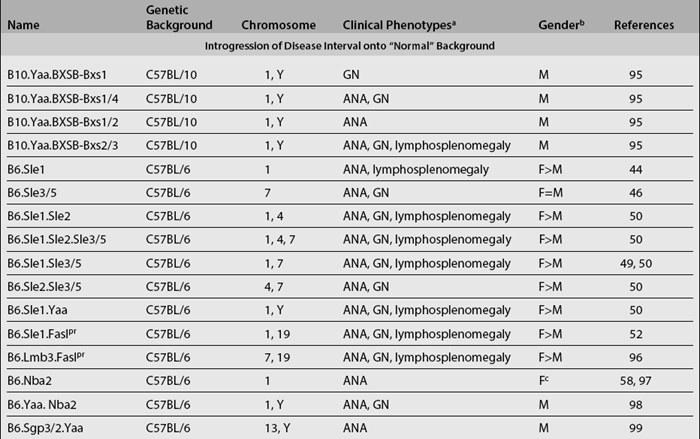
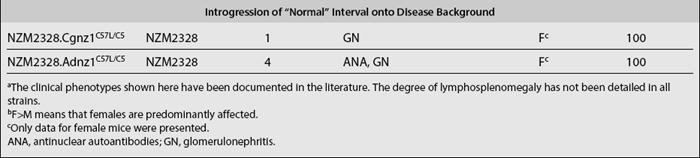
A good illustration of how lupus pathogenesis can be “dissected” using congenics stems from the genetic studies of the NZM2410 inbred model. In this model, lupus is contingent upon at least three non-MHC chromosomal intervals—Sle1, Sle2, and Sle3/5. Functional analyses of B6-based congenic strains bearing Sle1, Sle2, or Sle3/5 have demonstrated that each interval is responsible for very different component phenotypes.44–48 Among them, the breach of immune tolerance to nuclear antigens mediated by Sle1 appears to be essential for lupus development.12,49,50 However, Sle1 by itself does not lead to the development of fatal lupus but only modest serologic autoreactivity.49,50 In contrast, Sle1 mediates highly penetrant fatal glomerulonephritis in epistasis with Sle2, Sle3/5, Yaa, or lpr.49–53 Studies of the NZM2410-derived susceptibility intervals support a multi-step pathogenesis model, in which development of fatal lupus appears to be “initiated” by Sle1 and further exacerbated by additional susceptibility loci or genes.4,54
Congenic recombinant studies have identified that Sle1 is a cluster of four susceptibility subintervals: Sle1a, b, c, and d.55 Fine mapping of the Sle1b interval has already helped identify the association of extensive polymorphisms in the SLAM/CD150 gene cluster with the development of systemic autoimmunity.56 Likewise, the culprit gene responsible for the Sle1c locus appears to be polymorphisms of the CR2 gene.57 In the NZB model, Ifi202 has been implicated as the responsible candidate gene within the Nba2 locus on chromosome 1.58 Besides these reports, the culprit genes responsible for the other lupus susceptibility loci still remain elusive.
Congenic strains have also served as an excellent approach to investigate how various susceptibility loci may interact to mediate lupus development in terms of the cellular and molecular cascades involved. Thus, for example, whereas Sle2 appears to be responsible for the B-cell hyperactivity and the expanded B1 cells noted in lupus, Sle3 appears to be responsible for the hyperactive and proinflammatory antigen-presenting cells including dendritic cells (DCs) and macrophages.45,59–61 In contrast, Sle1 appears to be a gene that is very important in breaching B-cell and T-cell tolerance to self-antigens. However, the congenic dissection strategy still has its limitations. Although it is very powerful for segregating the disease susceptibility loci, it is much less so for further separating multiple disease genes that may be located within a small genomic interval. Identifying the phenotypic effect of each individual candidate gene within the small interval requires expensive genetic manipulation to generate engineered mouse models carrying individual candidate genes on a resistant-strain background in order to ascertain each gene’s contribution to disease. Nevertheless, this appears to be the most promising approach currently available to lupus researchers.
ENGINEERED MODELS CONSTITUTE POWERFUL TOOLS FOR STUDYING POTENTIAL ROLE OF INDIVIDUAL GENES IN LUPUS PATHOGENESIS
Studies of various genes using the knockout technology have revealed that deficiency in many proteins can lead to pathology similar to SLE (Table 17.3). These proteins have diverse functions, and the occurrence of lupus-like disease cannot always be predicted from their known functions at the outset of the studies. The implicated molecules thus far have been in various pathways that regulate the immune response. One class of proteins implicated in lupus mediates the clearance of apoptotic cells. This clearance may be essential for removing potential autoantigens that might otherwise serve as triggers of ANA production. These include, among others, serum amyloid P component (SAP), Dnase I, C1q, and c-mer. Among them, SAP specifically binds chromatin of apoptotic cells and nuclear debris, displaces H1 histones, and solubilizes native chromatin.62 Dnase I removes DNA from soluble or deposited autoantigenic nucleoprotein complexes.63 C1q also participates in the clearance of apoptotic cells.64 Disruption of the genes for these proteins apparently results in the impaired clearance of apoptotic cells. Consequently, the availability of excess autoantigens increases and apparently immune tolerance cannot be effectively maintained. Among these genes, C1q deficiency has been found to be associated with severe SLE in humans.65–68 Deficiencies in C2 and C4 also predispose to SLE.66,69–71 There is only one report on two SLE patients that show decreased DNAse-1 activity.72 Thus far, there have been no reports of deficiency of c-mer in human SLE. Similarly, C-reactive protein (CRP) appears to be genetically defective in some patients with SLE.73
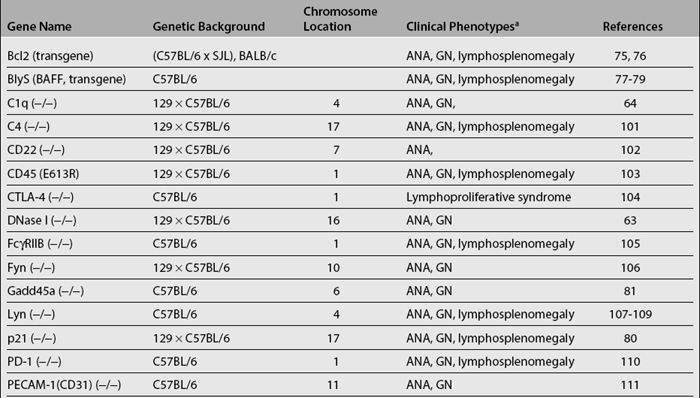
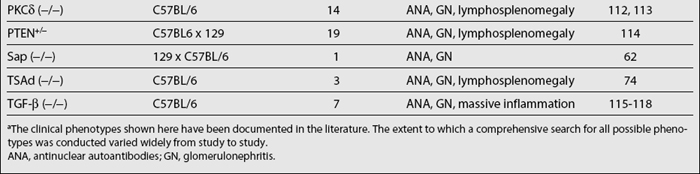
Proteins in a second group control B- and T-cell activation and proliferation. Most of these proteins are involved in cell signaling. It can be expected that disruption of any of the molecules crucial in negative feedback regulation of lymphocyte signaling could potentially lead to uncontrolled lymphocyte activation and possibly autoimmunity. The molecules in this category include Lyn, Fyn, CD22, TGF-beta, CTLA-4, PD-1, FcγRIIB, Pten (+/−), PECAM-1/CD31, PKC δ, and so on (Table 17.3). Genetic manipulations that promote lymphocyte activation and proliferation also lead to autoimmunity. T-cell—specific adapter protein (TSAd) has been implicated in regulating IL-2 production and T-cell apoptosis; the disruption of this gene results in defective T-cell death and development of systemic autoimmunity.74 Bcl2 and BLyS promote the survival of lymphocytes, and their overexpression also leads to the development of lupus-like autoimmune phenotypes.75–79 Disruption of molecules that are involved in cell-cycle checkpoints, such as Gadd45α and P21, can also precipitate autoimmunity.80,81 Among these genes, the association of specific genetic polymorphisms in PD-1, FcγRIIB, and BLyS with human SLE has been documented.82–84
Gene knockout technology represents a very powerful tool for studying the roles of individual genes in the development of autoimmunity. However, when interpreting results from models generated by this approach, limitations must be recognized. First, most of these knockout mouse models have been generated on the 129-strain background, and accumulating evidence indicates that this genetic background possesses several autoimmunity-promoting loci.85 It is even more critical when the targeted gene lies within a susceptibility interval, such as within distal chromosomes 1. Moreover, the knockout approach cannot provide much insight into elucidating the phenotypic consequences of subtle polymorphisms in candidate genes. In this regard, an ideal model would express the particular allele of the polymorphic gene on a resistant genetic background in order to help researchers fathom how subtle sequence differences can influence gene function.




