CLASSIFICATION
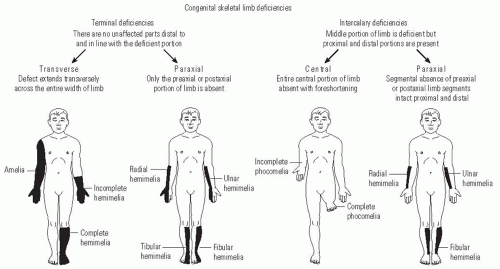
The attempt to classify congenital anomalies has evolved over time to more precisely describe each patient’s particular anomaly. The first descriptions of limb deficiencies used terms to describe specific phenotypes, such as “phocomelia” (phoke = seal) to describe the loss of the arm and forearm with attachment of the wrist and hand to the trunk or “lobster-claw hand” to describe the loss of central digits of the hand. A widely used classification system in the United States for congenital anomalies was devised by Frantz and O’Rahilly (
1) (
Fig. 30-1). This system differentiates between complete limb absence (amelia) and partial limb involvement. Partial limb involvement can affect roughly half of the limb (hemimelia), foot (podos), hand (cheir), digits (dactylos), or phalanges (phalanx). Deficiencies can be transverse, where the distal part of the extremity is lost and the proximal part is relatively normal; longitudinal, where one side of the limb (either the preaxial, postaxial, or central portion) is affected; and intercalary, where the proximal and distal limb are relatively unaffected with an intervening affected segment (
Table 30.1).
An international collaboration by the International Standards Organization (ISO) and the International Society for Prosthetics and Orthotics (ISPO) produced a classification system accepted as the universal language of national organizations that treat these children (
2). Many of the concepts of Frantz and O’Rahilly are incorporated into this system, although the Greek word roots have been eliminated. This system also uses the concept of transverse and longitudinal deficiencies. In the transverse deficiencies, the part of the segment at which the limb is missing is named, and the extent within that segment may be stated. Thus, complete limb loss at the midtibial level would be “transverse lower leg mid third.” In a longitudinal deficiency, the bone or bones missing are named from proximal to distal and described as “partial” or “total.” Therefore, a complete tibial deficiency with a hypoplastic great toe would be “longitudinal tibia complete, ray 1 partial.”
EPIDEMIOLOGY AND ETIOLOGY
Pediatric limb deficiency is uncommonly encountered by most pediatric orthopaedic surgeons. Depending on which global location is being considered, either congenital or acquired deficiencies may predominate. Congenital deficiencies are more common in developed nations (
3), while traumatic amputations can predominate in lesser developed nations (
4). Tumors are an uncommon but important cause of limb deficiency in children in all locations. Because limb deficiency is uncommon, patients are often treated in tertiary organized programs that have the experience and resources to treat these patients.
Depending on the methods used, the calculated incidence may vary widely, from 6 per 10,000 in British Columbia (
5) to 310 per 10,000 in Tayside, Scotland (
6). In a survey of European countries participating in the International Clearing House for Birth Defects Monitoring Programme, the incidence was between 3.1 and 7.9 per 10,000 (
7). Statistics such as these are more accurately determined by well-collected birth registries and less accurately determined by surveys from prosthetic clinic medical records, which can overestimate incidence if the clinic is a tertiary referral center.
Fibular deficiency is the most common cause of long bone congenital limb deficiency, when considering that fibular deficiency often accompanies femoral deficiency. Femoral deficiencies are the next-most common, with an incidence between 1 in 50,000 and 1 in 200,000 live births. Femoral deficiencies include the spectrum of the congenital short femur with a stable hip joint and a knee without significant contracture to proximal femoral focal deficiency (PFFD). The prevalence of tibial deficiencies is far less than either fibular or femoral deficiencies and is reported to be approximately one per million live births.
The incidence of all upper extremity amputations is not precisely known but is thought to be more than lower extremity amputations and more often congenital and bilateral than acquired (
8,
9). The most common congenital upper extremity amputation is by far the transverse forearm (below-elbow) amputation, with radial longitudinal deficiency being the nextmost common. In reality, few pediatric orthopaedic surgeons, other than those working in a limb-deficiency program, will have much experience with these amputations. Although the physician should strive to understand the cause of a congenital amputation in all cases, most of the time no identifiable cause exists. Limb deficiencies can be caused in several ways, such as by environmental factors, genetic disorders, vascular anomalies (such as “the subclavian artery supply disruption sequence”), (
10) and amniotic bands.
The oldest and most commonly held etiology for congenital amputation in the past was the mechanical amputation of limbs by amniotic bands, or Streeter dysplasia. Streeter postulated that the bands caused an intrinsic defect in the growth of the fetal limb (
11). There is, however, evidence that amniotic bands can form a constriction around the developing limb that interferes with the growth of the limb. The resulting constriction can lead to any degree of damage, from a constriction band around a limb that is otherwise normal to a complete
transverse amputation. The previously developed limb has actually been recovered at the time of birth, indicating the mechanism (
12). Evidence suggests that most amniotic bands cause deficiency within the first month postconception, based on the fact that limbs and organs commonly affected together are located in close proximity in the embryonic, but not fetal, stage of development (
13). Most children with amniotic band syndrome additionally have either craniofacial abnormalities or other evidence of band formation.
Modern genetics has shown that the development of the limb is a complex phenomenon that requires the precise interaction of a large number of genes and their effects, which are described in
Chapter 1 and other review articles (
14,
15). Many of the proteins and growth factors that participate in this complex interaction have been elucidated (
16). Genetic causes of limb deficiency can include chromosomal abnormalities (trisomy 18 and radial longitudinal deficiency), as well as single-gene defects, which result in deficiencies that closely follow Mendelian genetic transmission patterns (tibial deficiency, cleft hand and foot, radial longitudinal deficiency). Even the so-called sporadic deficiencies are more common in families with a history of similar deficiencies. A recent study from the Medical Birth Registry of Norway showed that children born to a mother with a limb deficiency had a relative risk of 5.6% of having the same defect as the mother (
17). This is similar to the relative risk of clubfoot. These facts carry consequences for genetic counseling. Understanding the cause of the deficiency is important to the resolving of the guilt that parents will initially feel. The possibility of a transmissible defect is certainly something both they and their affected offspring will also need to know. For the physician, knowing the existence of medical comorbidities and the natural history of the syndrome is necessary for the care of the child.
The disruption of the subclavian artery and its blood supply to the tissues explains the overlap of many of the common orthopaedic conditions seen, for example, Poland syndrome, Klippel-Feil syndrome, Mobius syndrome, Sprengel deformity, and transverse limb deficiencies. There are several possible
mechanisms by which this disruption may occur. For more details, the reader is referred to an excellent review article (
18).
Teratogens are an uncommon cause of limb deficiencies. Although typically thought of as medications, there are several other categories of teratogens during pregnancy, including maternal illnesses, medical diagnostic procedures, or trauma. To establish if a particular exposing agent is a teratogen, it should exhibit a dose-response relationship with the defect in question, and it should exhibit a period of greatest sensitivity. Thalidomide, an antinausea medication used in pregnant women in the 1950s and more recently as a chemotherapy agent, caused typical limb deficiencies during a narrow window maternal exposure (between 40 and 44 days postconception). It remains one of the most well-defined teratogenic agents causing limb deficiency. Other drugs are known to affect limb morphogenesis, such as warfarin, phenytoin, and valproic acid. Phenytoin and misoprostol have been shown to affect the vascularity of a previously normally developed fetal limb (
19). Retinoic acid and its related metabolites have an effect on limb bud development and cause a wide range of limb malformations in experimental models, which are described in an excellent review article (
16).
Almost all of the potential causes of limb deficiency previously described can and do affect other organ systems, often in recognizable patterns. This is an important fact for the treating physician, who should perform a thorough examination for other abnormalities and any heritable genetic defect should be identified. A knowledge of syndromes with limb deficiencies will help the physician look for associated abnormalities and enlist the help of other relevant medical subspecialists when appropriate.
PSYCHOSOCIAL ASPECTS IN LIMB DEFICIENCY PATIENTS
The Parents.
When a child with a congenital deformity or congenital amputation first presents, it is the parents who will need the most attention. Although surgical treatment of the child’s deficiency is not urgent, the parent’s emotions and expectations of treatment understandably take on an urgent tone. Unless prenatal ultrasound detected the deficiency, their child’s situation is unexpected and begins a grief cycle not unlike that described by the Swiss physician, Elisabeth Kubler-Ross (
20). It is this dynamic that the physician, who is likely meeting the family for the first time, must negotiate. In general, the physician’s responsibility over the next several months is both to suggest treatment according to the best medical standards and to guide the parents through the grief cycle.
Parents will initially feel shock and helplessness, which can manifest as feelings of guilt. What did I take or do during my pregnancy to cause this? In many families, there will be a strong desire to know “why this happened.” It is a time of anticipated joy that has turned into a period of great stress for both the individuals and, often, the marriage. The initial patient visit is important in establishing a positive doctor-patient relationship by active listening first, then explaining in clear terms the nature of their child’s deficit. Obtaining a genetic consultation, even when the deficiency is not known to be hereditary or caused by teratogens, can be therapeutic in this regard, giving the parents additional reassurance.
After the initial shock, parents will have many questions about their child’s future. The parents probably have never known a child or an adult with an amputation and have no frame of reference to answer these questions. Although the physician can give lengthy answers to these questions, it is important not to give them too much information all at once. The best one can do during the first visit of the parents is to gain their confidence and give them realistic hope. Fortunately, there are usually no emergent decisions to be made, and there is time to help the parents answer these questions for themselves.
During this initial period, physicians need to be careful about what they tell the parents. In an effort to help the parents feel better, physicians can be tempted to offer false hope and mention treatments that are totally unrealistic. Physicians who do not know, or who do not wish to take on this role, should assure the parents that they are referring them to the best possible care rather than tell them not to worry and that medical science has amazing cures today.
Parents will often refuse a recommendation such as an amputation (
21). It is important for the treating physician to recognize the factors that affect their decision and to do his or her best to educate the parents. From the parents’ point of view, their child may have a near-normal-appearing foot and a limb that is only slightly shorter than the normal one. Likely, their child will have little difficulty walking, and the parents may not understand the progressive shortening that will develop with time. Along with these observations, they share the popular public belief that modern science can cure everything—next year, if not now. They have all heard of miraculous lengthening of limbs in the popular press, and more recently, the “successful” transfer of limbs. It can be difficult to align the parents’ expectations with reality.
After the initial visit, the next several months are a good time for the parents to meet children with similar deficiencies and to see what their child might actually be like in the future. The literature demonstrates that although parents benefit from the support of friends and health professionals, they do not receive the level of support they need (
22). This support is found by contact with other parents whose children have similar disabilities. Meeting families and children in a similar situation is comforting, makes the future seem more manageable, and is one of the most important parts of treatment for these children. In addition, the parents can see the various surgical options that might be recommended for their child. It can help them form realistic treatment expectations by seeing what is possible rather than reading and hearing second-hand from other sources, such as the lay press, Internet, or well-meaning friends. Because there is no need for intervention in the congenital deficiency for a few months to years, the parents have time to learn about their child’s problem.
After parents have seen other children with limb deficiency, it is a good time to revisit their child’s diagnosis and medical treatment plan. By this stage, parents have a lot of information and notions of treatment, and they need guidance to place their child in the correct context of the information. The relationship between the parents and the physician (as well as all members of the team) is important. In this regard, the first thing the parents must be made to understand is that all the decisions will be theirs. Empowering well-informed parents to seek realistic treatment solutions for their child is an essential part of the treatment process. The physician’s role is to educate the parents through repetitive explanation and answering parents’ questions to ensure they are indeed informed. It is often the other members of the care team, such as the therapist and prosthetist, that the parents will usually “hear” and retain more information from at this stage. Again, nothing helps like seeing other patients and parents.
Finally, the physician should constantly reinforce that the child with a limb deficiency is more normal than abnormal. During this first year, the parents need to resolve their disappointment and loss, accept the child, and see the potential for the future. As part of that process, they need to bond with the child and begin to view the child as an independent person. Most parents will begin to see their children this way with growth and development. Again, this process is facilitated by seeing other children with similar problems. One recommendation that often needs repeating to the parents is that children tend to acquire the fears of their parents and that supporting any activity the child expresses interest in will allow the child to develop his or her natural abilities to the fullest.
The Child.
The child with an amputation is essentially different from other children. Although this is evident at birth in the case of congenital amputation, the child will not understand this difference until much later. Children’s understanding of their disability is general and incomplete at 6 years of age, but within a few years, around the age of 8 or 9 years, they come to a much more complete understanding of their handicap (
23). Therefore, if parents have not discussed this with the child, they can expect the more difficult questions from their child to begin at around this age.
All children with disabilities are vulnerable to social isolation. This, in turn, can have negative effects on the development of self-esteem, body image, and the child’s identity, which are developed through interaction with parents, teachers, friends, classmates, and others. As children develop these interactions, the issue of “first appearance” becomes important because it serves as a clue to perceived personal characteristics and can be an obstacle to further healthy interaction. Children in peer groups tend to devalue those with physical differences, a factor that may greatly interfere with these relationships (
24). Parents especially understand this and fear for their child in this regard.
There has been a great deal of study on the unaffected child’s reaction to various limb deficiencies, showing that children prefer other unaffected children and that they dislike some limb deficiencies more than they dislike others (
25,
26 and
27). In addition, it is known that young adults show signs of anxiety when face-to-face with a person with a limb deficiency. However, there is some evidence that young children do not share their parents’ values toward various limb deficiencies when young, but between the ages of 6 and 18 years, they gradually develop values almost identical to those of their parents (
26). This would suggest that these values may be subjected to modification among young children and emphasizes that organized discussions with classmates in school about the child’s handicap may be of great value.
Despite the negative “first impression” that physical differences hold for children, there is evidence demonstrating that the age of the patient, the gender, the degree of limb loss, or socioeconomic status are not predictors of low self-esteem or of depressive symptoms. Rather, social factors, for example, stress and hassle, parental discord, and social support from classmates, parents, and teachers, along with the child’s own perceptions of competency and adequacy, gained through peer acceptance, scholastic achievement, and athletic accomplishments, play the largest role in the development of self-esteem (
28,
29 and
30).
The importance of this information for parents, physicians, therapists, prosthetists, and teachers is that although limb deficiency is the visible problem and is subject to little modification, the important factors in the development of self-esteem are independent of the deficiency and can be positively affected.
CARE COORDINATION
The management of pediatric limb deficiency is best undertaken in a multidisciplinary specialized clinic. There are several reasons for this. First, limb deficiency is rare, and tertiary limb deficiency centers allow participating caregivers to gain the necessary experience to successfully treat these patients. It is beneficial to have these caregivers in the same place at the same time. This makes it convenient for the parents to receive their child’s care and can facilitate the efficient transfer of information, both between treating professionals and parents as well as among different treating professionals. Second, as previously mentioned, all patients and parents benefit by knowing they are not alone. They will benefit immensely by seeing and speaking to other parents and children like themselves.
The team should be made up of a physician and surgeon, a prosthetist, a physical and occupational therapist, and a social worker or child psychologist, all of whom are knowledgeable in normal childhood development and who can anticipate the deviations that will occur in development. As compared to adult acquired amputees, who know what they had and what they want, the child and parents of a congenital amputee know little and need education and guidance that usually cannot be provided by an orthopaedic surgeon referring the patient to a prosthetist. The parents will be making decisions for their child with lifelong consequences, and they are acutely aware of
this responsibility. The professionals caring for the family must provide the necessary education and framework in which the parents can make these decisions.
Family involvement is an essential part of the treatment program (
31). The child has a condition he or she will adapt to, rather than a disease that can be cured. Hence, the condition should not be “medicalized,” but rather treated within the context of family, home, school, and play, not through clinic visits.
GENERAL TREATMENT PEARLS AND PITFALLS
Predicting Growth.
It has been observed that the percentage of shortening in a congenital limb deficiency remains relatively constant. This is sometimes referred to as the “rule of proportionality.” This principle has been established for congenital short femur (
32,
33 and
34) and fibular hemimelia (
35,
36). Clinical experience indicates this to be true for the tibial hemimelias also. It follows from the rule of proportionality that differences in limb length will increase as the child grows. Therefore, in discussing centimeters of shortening and planning treatment, it is important to calculate what the discrepancy will be at maturity rather than focus on what it measures currently. This can be roughly estimated by knowing only the percentile height of the child. With this information, the length of the femoral and tibial segments of the normal limb can be estimated from the Green and Anderson growth charts (
37) (
Tables 30.2 and
30.3). Then, knowing the length of the normal segments and the percentage by which the affected segments are short, the length of the affected segments at maturity can be estimated.
Although this method of calculating the eventual discrepancy at maturity is clinically valid, the clinician should be aware of the effect that surgical procedures could have on the growth of the limb. Following amputation, the epiphysis of the bone may not grow at the normal rate. Christi et al. (
38) showed that in 20 below-knee (BK) amputations in children, only three tibias grew at the expected rate. The congenital group of tibias grew to 36% of what would have been expected, and the acquired group grew to 53% of the expected level. This may be due to the lack of stress across the growth plate, the decreased blood flow to the bone, or the result of the congenital insult that produced the limb deficiency.
Timing of Amputation.
The timing of an amputation in a congenital limb-deficient child is tied into the developmental age of the child. In general, amputations for lower extremity congenital deficiency are elective and designed to aid prosthetic fitting. As such, amputation is best performed a few months before the child is developmentally ready to walk (usually when the child is pulling to stand). This gives enough time for the residual limb swelling to subside and for fabrication of the prosthesis. This will allow the child after surgical recovery to maintain a normal developmental sequence. In rare instances, a deformed extremity can interfere with crawling, and amputation may be performed earlier. However, prosthetic fitting in such children should wait until it will be of some functional value. In other cases (e.g., PFFD), amputation may be done at a slightly later age (and after prosthetic fitting because of technical reasons).
Although it is poorly documented, there is the impression among both parents and surgeons that with early amputation the child does not experience the body image loss that accompanies amputation at a later stage. Also important is that as a general rule, the earlier the amputation, the better the adaptation of the child’s neurologic plasticity to the alteration. No upper age limit has been identified, although most amputations should be performed before school age, if possible.
Overgrowth.
Bony terminal overgrowth at the end of the residual limb is the most common problem in juvenile amputees. Its occurrence is reported to be between 20% and 50% and depends on the cause of the amputation, the age of the patient at the time of amputation, the bone involved, and the location within the bone involved (
39,
40 and
41). It occurs most commonly following traumatic amputation or elective amputation through a bone. It is less often seen in congenital amputations because of amniotic band syndrome but not in those due to failure of limb development. It is not seen in amputations through joints (
42). Overgrowth occurs most often in below-knee amputations, with the problem being present in the fibula more often than in the tibia, and in transhumeral amputations. The incidence of overgrowth is less common if the primary amputation is performed before the age of 12 years. Recurrence is common and is felt by some to be more common during periods of rapid growth when bone turnover is high (e.g., adolescence).
Contraction of the soft tissue and physeal-mediated growth of the bone, pushing it through the skin, were originally thought to be responsible for bone overgrowth. Aitken disproved these theories when he demonstrated by implanting metallic markers that the overgrowth took place distal to the end of the bone (
39,
43). The new bone is periosteal and endosteal appositional bone. Overgrowth results from the typical process of wound contracture as has been demonstrated by Speer (
44). Following a through-bone amputation, the periosteum continues to grow. As it grows over the end of the bone, it grows over the open medullary canal, where it contracts and is drawn into the canal from which it can continue to grow, producing the overgrowth at the end of the bone.
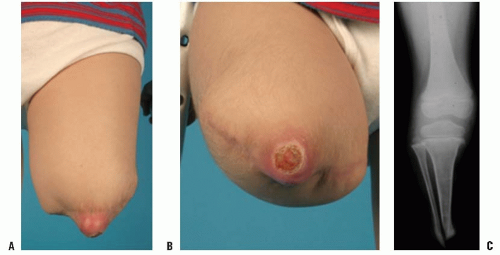
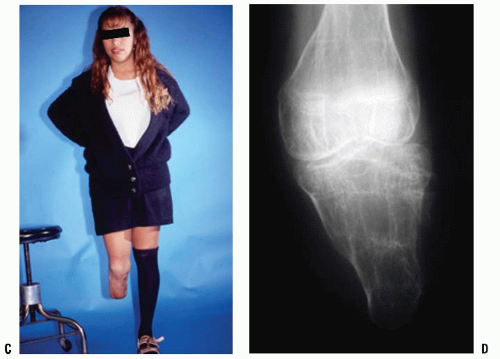
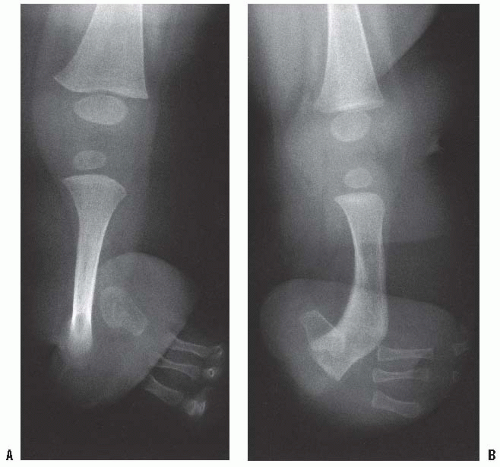
Patients with terminal overgrowth present clinically with pain on weight-bearing or prosthetic use. An antalgic gait with decreased stance time may be noticed. Decreased range of motion, to limit pulling of the skin at the end of the limb, is an additional symptom. Clinically, the patient presents with tenderness and pain at the end of the residual limb. There may be inflammation, bursal formation, or the bone end may be protruding through the skin. Commonly, the bony spike can be palpated within a small, tender bursa (Fig. 30-2A-C).
Routine evaluation can detect the problem before it is painful, and surgical correction is fairly straightforward at this point. Prosthetic adjustments may help delay surgical revision, but will seldom be sufficient with significant overgrowth.
Treatment of overgrowth has included implantation of various plastic and metallic devices over the end of the bone (the so-called stump-capping procedures). However, these techniques are not commonly used because the results are no
better than the use of biologic material (
45,
46). Marquardt reported in the mid-1970s, in the German literature, on the capping of the bone end with a cartilage-bone graft. More recently, Tenholder et al. (
47) reported comparable results with the use of a polytetrafluoroethylene felt pad. Various reports for both acquired and congenital amputees indicate generally favorable results, with most revisions being for technical reasons (
45,
46,
48,
49).
When a patient presents with pain and bursa formation, prosthetic modification and other conservative measures
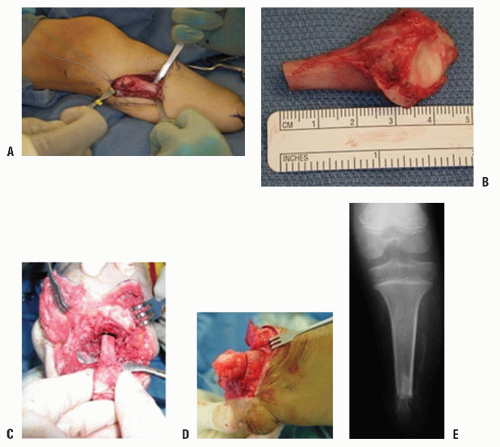
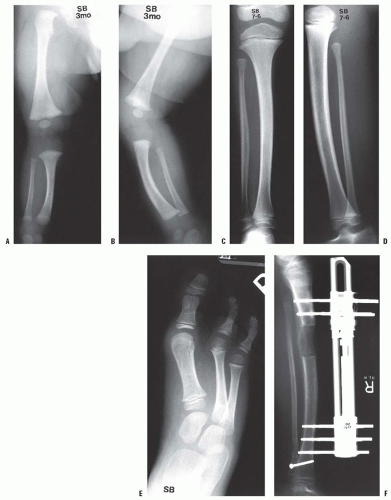
should be used first unless a bony spike is felt. If revision surgery is necessary, a capping procedure should be considered. In the case of a primary amputation, it is advisable to use available parts from the amputated portion of the limb to cap the end of the bone, if conditions permit. The most common procedure is the use of the proximal fibula to cap the tibia (
Figs. 30-3A-E). As in any revision, adequate resection of the bone is essential to provide a healthy soft-tissue envelope. Harvest of the proximal fibula involves detaching the lateral collateral ligament of the knee, which can theoretically lead to lateral knee instability. However, there have been no reports of lateral knee instability after proximal fibular resection for biologic capping, and the literature regarding knee instability after proximal fibular resection for tumors is mixed (
50,
51,
52,
53 and
54). Given that the literature is unclear on the need for lateral ligamentous reconstruction, it seems reasonable to test intraoperative knee instability and repair or reconstruct the ligament if necessary.
Following surgery, the therapist should supervise and educate the child and parents in edema control to accelerate return to prosthetic use. In addition, range-of-motion and strengthening exercises accelerate, and may even be necessary to regain, the full function of the prosthesis. Users of myoelectric prostheses may need readjustment of their electrodes and, for a while, may have difficulty activating the prosthesis. This is because swelling and reshaping of the limb may alter the optimal sites for electrode placement.
Short Residual Limb.
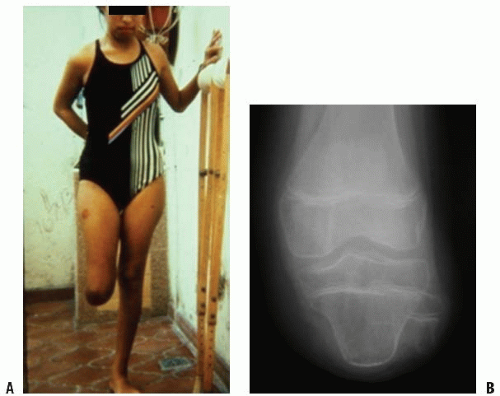
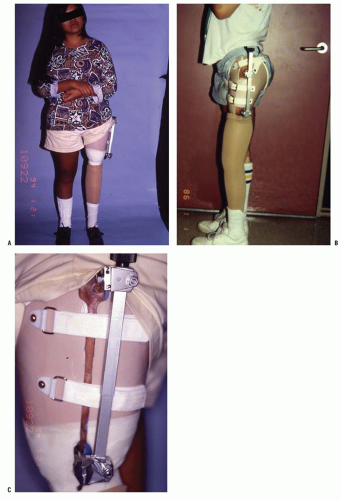
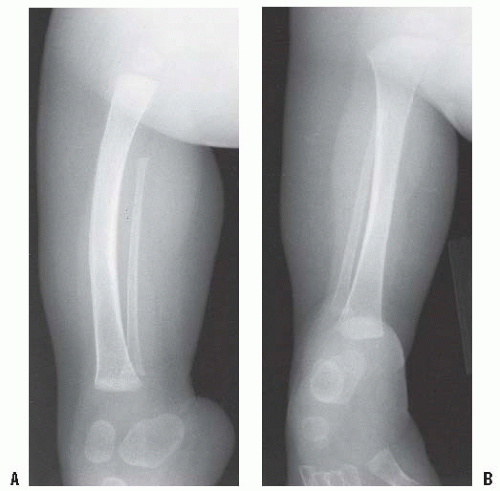
In some patients with either congenital or acquired amputation, the residual limb will be too short for satisfactory or comfortable prosthetic fitting. In such cases, it may be possible to lengthen the residual limb. Watts has written an excellent review of this subject. Favorable results have been reported for both upper and lower extremity residual limb lengthening (
55,
56) (
Figs. 30-4A-D). Alekberov et al. (
57) report on six patients who had successful lengthening of 3.4 to 8.4 cm in congenital below-elbow segments. The remainder of the literature to date consists largely of case reports.
The lengthening of residual limbs is fraught with complications, and careful consideration needs to be given to the potential benefits versus the possible complications. Deficient soft-tissue coverage is the main limit to adequate lengthening. Tissue expanders generally do not provide the solution. Free tissue flaps may be used when skin coverage is inadequate. Free flaps often remove sensation from the end of the residual limb, and especially in the upper limb this can affect the function of the limb both with and without the prosthesis. Although difficult, it is possible to fabricate a temporary prosthesis for use during the lengthening process for both above- and below-knee prostheses so that the child can continue weight bearing, at least during the consolidation phase (
55) (
Fig. 30-5A-C).
Phantom Sensation/Pain.
In the middle of the 19th century, the neurologist Silas Weir Mitchell coined the term “phantom limb.” He described sensations as replicas of the lost limb, some being painful and some not. Phantom sensation of the limb is often described by patients as the feeling that they can move the part, tell how the part is positioned, or feel it itching or tingling. Phantom pain, however, is perceived by the patient as just that—painful. It often is the same as the pain before an amputation or may be cramping, shooting, burning, or of any other characterization.
It was generally thought that children born without limbs do not have sensations of them; nor do these children experience the phantom pain or phantom sensation seen in the acquired amputee (
58). Recent reports call this commonly accepted truism into question (
59,
60). Whatever this pain is, those who care for children with limb deficiencies will recognize that these children do not have the same problem as the true chronic phantom pain seen in adult amputees.
Melzack et al. (
60) reported that at least 20% of children with congenital limb deficiency and in 50% of those who underwent amputation before age 6 experienced phantom limb. Of those, 20% of the congenitally deficient group described the sensations as painful, whereas 42% of the acquired amputees described them as painful. To explain the phenomenon of phantom limb in a child who has never had a limb, Melzack et al. have proposed that there is a genetically or innately determined neural network that is distributed in the cortex (not focal), which is responsible for the representation
of the limb, even in the absence of normal limb bud development.
Phantom pain and distal residual limb pain are also commonly associated with other pains, such as headache, bone, or joint pain (
59,
61). The frequency of phantom sensations varies. They are often triggered by a wide variety of stimuli. Feeling nervous or happy, not wearing a prosthesis, being cold, or being ill are frequent triggers. Fortunately, these sensations do not interfere with the child’s usual activity, and most children say they just try to ignore the sensations (
62).
There is good evidence in adult patients that preemptive analgesia during amputation surgery, or immediately in the postoperative period, can decrease postoperative phantom pain in adults, and it has been suggested that the same is true in children (
63). Epidural or spinal anesthesia can decrease postoperative limb pain as compared to general anesthesia (
64,
65). Postoperative continuous-infusion intraneural catheters have also been used with success (
66,
67). For established phantom limb pain, there is no single highly successful treatment. Because many of these problems resolve with prosthetic alterations or physical therapy modalities, a multidisciplinary approach has proven to be the best intervention in evaluating and properly treating the phantom limb phenomenon when it becomes a problem.
A properly fitting socket, with appropriate suspension and sock thickness, is the best and first treatment of choice (
62,
68). A heavy, tight shrinker, either worn inside the prosthesis or when the prosthesis is off, may provide relief. Physical therapy interventions, including weight-bearing and graduating pressures such as tapping, rubbing, and massage to the residual limb, have been reported to give temporary or permanent relief. Rubbing and massaging the uninvolved limb at similar points to those in which they are experiencing the phantom limb sensation may provide relief. Various physical modalities have been utilized in the treatment of phantom sensations in children, including transcutaneous electrical nerve stimulation, biofeedback, ultrasound, and the physical agents of heat and cold (
69).
For the occasional adolescent amputee who has problems with phantom pain following an amputation, gabapentin (Neurontin, Park-Davis) has proven a useful medication for some patients (
70).