Chapter 10 T-Cells and Systemic Lupus Erythematosus
INTRODUCTION AND BACKGROUND
T-cells appear to play a central role in the complex series of events that culminates in the development of SLE. In this chapter, we consider loss of immunologic tolerance, autoantigen reactive T-cells, abnormalities of T-cell signaling, autoantigen processing and presentation, abnormalities of T-cell co-stimulation, and the role of T regulatory cells in disease pathogenesis. A model of how these factors may influence disease pathogenesis is shown in Fig. 10.1.
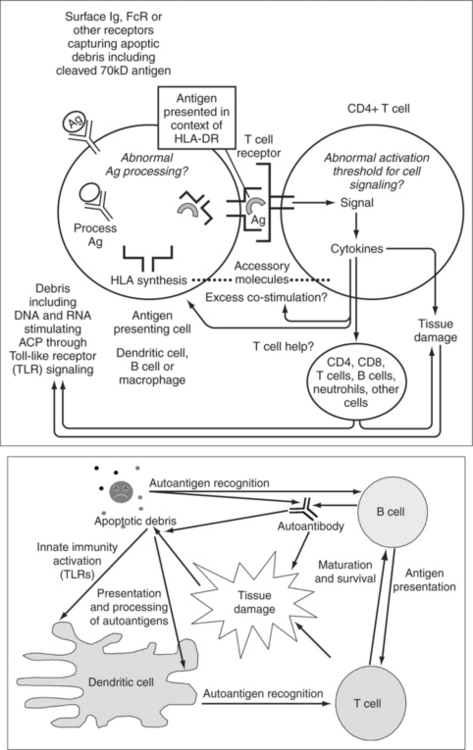
It is well established that T-cells are a central component of the adaptive immune system, and numerous observations support a role for T-cells in SLE pathogenesis.1–4 Early descriptive studies reported (and subsequent studies have repeatedly confirmed) that T-cells obtained from patients with active SLE display markers of activation (such as increased expression of HLA-DR on the cell surface2) and exhibit gene expression typical of cellular activation, including up-regulation of proto-oncogenes such as c-myc.5 Histologic studies have demonstrated close physical proximity of activated T-cells to sites of tissue injury. Studies have also documented infiltration of affected organs, such as the salivary glands and kidneys with T-cells.4,6–8 A recent example was a study using renal biopsy in SLE patients where it was found that infiltrating T-cells in the kidneys can be oligoclonal based on restricted T-cell receptor usage. These T-cells also exhibit evidence of recent activation.7
Indirect evidence of a role for T-cells in the pathogenesis of SLE has been suggested by immunogenetics studies where associations between select major histocompatibility complex (MHC) alleles and SLE have been reported.9 These studies have been interpreted as supporting a central role for T-cells in SLE pathogenesis, either through direct effects on T-cell-peptide-MHC antigen selection and presentation or through indirect effects on T-cell repertoire selection. Furthermore, in support of such a hypothesis antinuclear antibody-producing B-cells exhibit evidence of having undergone T-cell-derived cytokine-driven affinity maturation. Such B-cells are of the IgG isotype, and this IgG can be present in high levels in serum.8 Immunoglobulin heavy and light chain gene rearrangements in autoantibody-producing murine B-cells, and in the more limited number of instances where they have been examined in SLE patients, demonstrate nucleotide substitutions typical of an antigen-driven T-cell-dependent immune responses. These studies indirectly support a central role for T-cells in disease pathogenesis.
LOSS OF IMMUNOLOGIC TOLERANCE IN SLE
To participate in the pathogenesis of SLE, T-cells presumably must escape normal immunologic tolerance. There are a number of possible mechanisms whereby immunologic tolerance may be lost by T-cells, leading to autoimmunity. Some of the proposed mechanisms are summarized in Table 10.1.
TABLE 10.1 IMMUNE TOLERANCE: PROPOSED DEFECTS IN CENTRAL OR PERIPHERAL TOLERANCE IN SLE
| Central tolerance: |
| Peripheral tolerance: |
It has been demonstrated that T-cells reactive with self-antigens can escape thymic selection and are detectable in the peripheral blood of both SLE patients and healthy individuals.10–16 The T-cell receptor on these T-cells has low avidity for self-antigens and this may in part be why they are able to escape deletion in the thymus. In healthy individuals, such self-reactive T-cells are presumably kept from expanding by regulatory mechanisms. For example, during T-cell activation in healthy individuals such autoreactive T-cells may become anergic, undergo activation-induced cell death, or be limited in their expansion by other regulatory cells. Under the pathologic conditions found in SLE, however, autoreactive T-cells persist and expand and thereby contribute to the development of disease. Theoretical mechanisms for the expansion of such pathogenic T-cells include stimulation by newly exposed cryptic T-cell epitopes on self-antigens during apoptosis or during antigen processing, inappropriate T-cell help, excess T-cell co-stimulation, abnormal activation thresholds for T-cell signaling pathways, and inadequate control of T-cell growth by regulatory cells. The evidence for each of these in SLE is considered in this chapter.
AUTOANTIGEN-REACTIVE T-CELLS
Self-reactive T-cells may escape normal mechanisms of immunologic tolerance, expand, and be detectable in the peripheral blood of patients with SLE.10–16 T-cells reactive with a number of lupus nuclear autoantigens [including DNA; histones; the small nuclear ribonucleic proteins Sm-B, Sm-D, U1-70kD, and U1-A; and heterogeneous nuclear ribonucleoprotein (hnRNP) A2 protein] have been isolated from the peripheral blood of SLE patients and characterized. These are outlined in Table 10.2.10–12,17–32
TABLE 10.2 ANTIGEN-REACTIVE T-CELLS




Rajaogopalan et al. were the first to describe human T-cell lines reactive with double-stranded DNA isolated from patients with SLE.17 The activated T-cells they identified selectively augmented the production of pathogenic IgG anti-DNA antibodies ex vivo, supporting the conclusion that they might have a role in pathogenesis.17 Datta and colleagues subsequently characterized chromatin-reactive T-cells in SLE in detail and reported that these T-cells are typically CD4+, can provide help to anti-DNA and antihistone antibody-producing B-cells, and that they have restricted T-cell receptor CDR3 usage with characteristics of antigen selection by a limited number of cationically charged antigenic epitopes. They mapped the major T-cell epitopes present on the core nucleosomal histone protein complex to four regions: histone H2B amino acid residues 10 through 33, histone H3 residues 85 through 105, histone H4 residues 16 through 39, and histone H4 residues 71 through 94. They demonstrated that these autoantigenic peptides can be promiscuously presented by several HLA-DR alleles. Furthermore, they found that nucleosome-reactive human T-cells produce substantial quantities of INF gamma. They found in parallel studies done in a murine model system that such nephrogenic complement-fixing antinucleosome autoantibodies belong to INF gamma–dependent IgG subclasses. They subsequently proposed that expansion of these low-affinity chromatin autoantigen-reactive T-cells is essential for sustaining anti-DNA/histone autoantibody-producing B-cells.17–19
In addition to DNA and nucleosomes, human T-cells reactive with various small nuclear ribonucleoprotein self-antigens (including Sm-B, Sm-D, U1-70kd, and U1-A) have been identified and characterized. The characteristic features of autoantigen-reactive T-cells that have been described are outlined in Table 10.3.10–12,20,21,23–32 These small nuclear ribonucleoproteins are ubiquitous self-antigens that are components of the spliceosome complex, which physiologically functions to excise introns and generate messenger RNA transcripts lacking intervening RNA.33,34 Sm-reactive T-cell lines and T-cell clones reactive with the Sm-D or Sm-B small nuclear ribonucleoproteins were first described by Hoffman and colleagues from patients with SLE.10 U1 small nuclear ribonucleoprotein-reactive peripheral blood T-cells were first reported by O’Brien and colleagues from patients classified as SLE.20 T-cell clones from connective tissue disease patients reactive with the U1-70kD small nuclear ribonucleoproteins antigen were described by Hoffman and colleagues.23,26 Okubo and colleagues were the first to describe peripheral blood mononuclear-cell-derived CD4+ T-cells from SLE or MCTD patients that reacted to the U1-A small nuclear ribonucleoprotein.21 Subsequently, such various small nuclear ribonucleoprotein-reactive T-cell clones have been extensively characterized (see Table 10.3). Typically, they are CD4-positve T-cells that produce large amounts of IFN gamma, moderate quantities of IL-2, and variable quantities of IL-4 and IL-10.4,23–30 They recognize antigen in the context of HLA-DR. T- and B-cell responses are linked in SLE, and both small nuclear ribonucleoprotein and hnRNP-reactive T-cells can provide B-cell help for autoantibody production.31
TABLE 10.3 CHARACTERISTICS OF AUTOANTIGEN-REACTIVE T-CELLS
T-cell epitope mapping studies of human T-cell clones reactive with the small nuclear ribonucleoproteins U1-70kD, Sm-B, and Sm-D have been done to determine the precise regions recognized on the autoantigen by T-cells. These studies have revealed that there are limited T-cell epitopes on these antigens. Interestingly, virtually all T-cell antigen recognition regions (or so-called T-cell epitopes) reside within functional regions of the protein—either within the Sm motifs for Sm-B and Sm-D or within the RNA binding domain for U1-70kD and hnRNP.27,29,34
T-cell clones have recently been identified and characterized from patients with SLE that are reactive with another nuclear ribonucleoprotein antigen known as hnRNP A2.31 Greidinger and colleagues cloned human T-cells reactive with hnRNP A2 from SLE patients and found that such hnRNP-reactive T-cells when cocultured in vitro with autologous B-cells could augment anti-hnRNP autoantibody production.31 Haffman and Steiner also identified and characterized hnRNP-reactive T-cells and found that similar to the findings described previously for U1-70kD T-cell epitope mapping, hnRNP-reactive T-cells also recognize the RNA binding domain portion of the antigen.32 Collectively, these studies reveal a recurring theme: ribonucleoprotein-reactive T-cells are directed against highly conserved regions that function to bind their associated RNA (the RNA binding domain of U1-RNP and hnRNP) or their associated proteins (Sm protein-protein binding domains).
Finally, a novel mechanism for autoantigen “cross reactivity” by T-cells in SLE has recently been reported. De Silva-Udawatta and colleagues reported that T-cell receptor usage by small nuclear ribonucleoprotein-reactive T-cells can have significant flexibility or “plasticity”.30 For example, they found that a single T-cell receptor can recognize two distinct small nuclear ribonucleoprotein autoantigenic peptides that have no apparent sequence homology.30 This cross reactivity is limited to the U1-70kD and a Sm-B peptide. However, a series of other closely related small nuclear ribonucleoprotein-derived peptides did not cross stimulate the T-cell receptor. These studies indicate that there are now a number of distinct mechanisms for immunologic cross reactivity that may result in loss of tolerance in SLE, including cross reactivity occurring at the level of the T-cell receptor.




