Chapter 9 T Cells
Extensive evidence indicates that T cells are involved in the pathogenesis of systemic lupus erythematosus (SLE).1 The phenotype of T cells isolated from patients with SLE is abnormal: SLE T cells partially resemble activated cells and partially behave like anergic (unresponsive) cells.2 Their response to stimulation through the T-cell receptor (TCR) is exaggerated,3 and their gene expression profile is altered in comparison with cells obtained from healthy individuals.4 Moreover, the tolerance breach and self-directed response developed by patients with SLE have all the characteristics of a T-cell–driven immune response, including clonal expansion and somatic hypermutation,5 and T-cell depletion prevents lupus in murine models.6 Thus, even though SLE is a complex disease caused by multiple factors, evidence supports the role of T cells as promoters of the pathologic autoimmune response and as direct instigators of target organ damage. The aim of this chapter is to discuss the mechanisms by which T cells contribute to SLE and the intrinsic abnormalities that alter the behavior of the SLE T cells contributing to their pathogenicity.
Role of T Cells in Autoimmunity and Inflammation
Help to B Cells
CD4+ T cells regulate production of antibodies in germinal centers (GCs), specialized lymphoid structures where B-cell proliferation and differentiation occur simultaneously with isotype switching and somatic hypermutation. In normal immune responses, these processes ensure the selection of high-affinity antibodies and memory B cells. In patients with SLE, the response to autoantigens and the development of high-affinity autoantibodies are poorly understood. However, the fact that the autoantibodies are mostly high-affinity immunoglobulin (Ig) G that have undergone somatic hypermutation suggests that they have developed in GCs or analogous structures.7 Moreover, the presence of certain activated B-cell subsets in the peripheral blood of patients with SLE has been proposed to reflect increased GC activity,8 probably related to increased CD40 ligand (CD40L) expression.9
Follicular T-helper cells (TFH cells) represent the CD4+ helper subset specialized in providing help to B cells in GCs (Figure 9-1)10 TFH differentiate from naïve CD4+ T cells activated in the presence of interleukins IL-6 and IL-21 and co-stimulation through the co-stimulatory molecule ICOS. TFH cells localize to lymph node B cell zones and induce isotype switching and somatic hypermutation through IL-21 and CD40L.10 Their pathogenic capacity was shown in mice deficient in the ubiquitin ligase roquin. These mice demonstrated an expansion of TFH cells and systemic lupus-like autoimmunity.11 On the other hand, a mutation that disrupted the capacity of regulatory CD8+ T cells to suppress TFH cells also triggered autoimmunity.12 IL-21 and TFH cells have been shown to play a role in disease development in murine models of lupus.13–15 In lupus-prone B6-Yaa mice, defective CD8 regulatory function was associated with increased TFH activity and systemic autoimmunity.16 In MRL/lpr mice, deficiency of ICOS was protective because of the loss of extrafollicular T helper cells, an analogous CD4+ subset that promotes antibody production in extrafollicular compartments in autoimmune mice.13 A subset of patients with SLE was shown to have increased numbers of TFH cells in the peripheral blood, suggesting that indeed overactivation of this T-cell subset could be involved in human SLE.17 The aforementioned studies, along with the presence of high titers of high-affinity IgG autoantibodies in most patients with lupus, indicate that T-cell–driven B-cell hyperactivity is an key event in the self-directed immune response that underlies pathology in patients with SLE.18
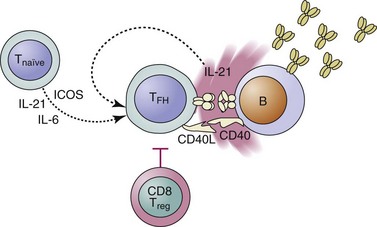
FIGURE 9-1 T follicular helper cells (TFH) are the CD4 subset specialized in providing help to B cells.
Production of IL-10, another cytokine able to promote B-cell function, is increased in patients with SLE.19 In certain populations, high IL-10 production has been associated with polymorphisms of the Il10 gene. IL-10 stimulates B cells and promotes immunoglobulin production by mononuclear cells of patients with SLE.20 Interestingly, in patients with SLE most IL-10 derives from B cells and monocytes. Blockade of IL-10 delayed disease in NZB/NZW mice21 and led to joint and skin disease improvement in a small group of patients with SLE,22 suggesting that it may indeed play a role in disease pathogenesis.
Promotion of Inflammation
Inflammation is controlled locally by the regulation of vascular permeability and tissue access to immune cells. T cells from patients with SLE produce large amounts of proinflammatory cytokines and express high levels of adhesion molecules. A 2011 study highlighted the prognostic importance of kidney tubulointerstitial infiltrates in patients with SLE, emphasizing the relevance of target organ infiltration by immune cells.23
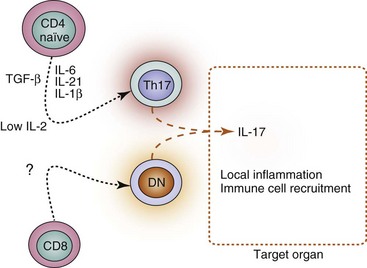
Th17 cells are a cell subset generated from naïve CD4+ T cells activated in the presence of transforming growth factor beta (TGF-β) and certain proinflammatory cytokines, including IL-1β, IL-6, and IL-21.24 Th17 cells induce inflammation through the release of IL-17A, IL-17F, and IL-22. Abnormally high levels of IL-17 are found in the serum of patients with SLE.25 Accordingly, abnormally high numbers of T cells produce IL-17 in patients with SLE.26,27 In lupus, however, Th17 CD4+ cells are not the only relevant source of IL-17, because expansion of a normally rare IL-17–producing T-cell population that lacks CD4 and CD8 (hence called double-negative, DN) is common.26 The heightened production of IL-17 in patients with SLE correlates with disease activity.25 Further, IL-17–producing T cells have been found within kidney infiltrates from patients with lupus nephritis.26
IL-17 production is also increased in murine models of lupus. Spleen cells from SNF1 mice produce high amounts of IL-17 in the presence of nucleosomes.28 As in patients, IL-17–producing T cells have been found in kidneys of mice with lupus-like nephritis.28,29 Interestingly, disease amelioration was accompanied by a reduction of IL-17 production in two murine studies.28,30
Different factors could account for the increased production of IL-17 in patients with SLE (Figure 9-2). Abundance of the Th17-promoting cytokines IL-6 and IL-21,31 along with reduced levels of IL-2,32 which promotes regulatory T-cell differentiation and inhibits Th17-cell generation,33 could skew the CD4+ T-cell priming. On the other hand, expansion of DN T cells increases the frequency of IL-17–producing cells.26 Thus, underlying inflammation, as well as lupus-specific factors (such as low IL-2 production), probably skews the effector differentiation of CD4+ and CD8+ T cells toward proinflammatory subsets that release cytokines that amplify the autoimmune response.
Interferon gamma (IFN-γ) is a proinflammatory cytokine produced by Th1, CD8+, and natural killer (NK) cells. Although some reports argue that IFN-γ production is decreased in patients with SLE,34,35 this decrease has not been uniformly documented.27,36,37 Importantly, IFN-γ–positive cells and IFN-γ RNA transcripts have been found in glomeruli from kidneys affected by lupus nephritis.38,39 In murine lupus models (e.g., NZB/NZW and MRL/lpr), IFN-γ plays a demonstrated pathogenic role.40,41
T cells are guided by adhesion molecules into lymphoid organs or peripheral tissues. CD44 is an adhesion molecule that binds to hyaluronic acid and other components of extracellular matrix. Its expression is increased on T cells from patients with SLE.42 Moreover, the affinity of CD44 is enhanced in SLE T cells, allowing increased migration of T cells into inflamed organs.42,43 The CD44 gene can yield variant isoforms through alternative splicing. The expression of two variants, CD44v3 and CD44v6, is increased on T cells from patients with lupus and correlates with disease activity, renal disease, and anti–double-stranded DNA (anti-dsDNA) antibody production.44 The relevance of this finding is exemplified by the fact that T cells that infiltrate kidneys in patients with lupus nephritis express CD44v3 and CD44v6.45
CD8+ and Double-Negative T Cells
The phenotype and function of CD8+ T cells has been scarcely studied in patients with SLE. Activity of peripheral blood CD8+ T cells may be increased in patients with active SLE,46 because a higher fraction of these cells express perforin and granzyme B.47 Importantly, CD8+ T cells are also found within cellular infiltrates in kidney biopsy specimens from patients with SLE, particularly in the interstitial and periglomerular areas,48 suggesting that the cells may participate in target organ tissue injury. On the other hand, cytotoxic capacity of CD8+ cells has been reported to be hampered in SLE.49
Although scarce in healthy individuals, DN T cells constitute a significant proportion of T cells in patients with SLE.26,50 When expanded, DN T cells probably play a pathogenic role in patients with SLE. They can provide B-cell help,50,51 produce proinflammatory cytokines (e.g., IL-1β, IL-17, and IFN-γ), and are found in cellular infiltrates in kidneys of patients with lupus.26 At least a fraction of DN T cells is derived from activated CD8+ T cells.52 In SLE, DN T-cell expansion is probably explained by either increased conversion of CD8+ T cells into DN T cells or abnormal survival of the latter.
Regulatory Function
T cells also suppress immune responses. This function allows the duration and intensity of the immune response to be controlled. Moreover, it protects the host tissues from immune-mediated damage.53 Complete absence of regulatory T (Treg) cells causes a severe autoimmune disorder in mice and humans (immunodysregulation polyendocrinopathy enteropathy X-linked syndrome [IPEX]).54 On the other hand, partial defects in numbers and function of Tregs have been linked to several autoimmune disorders, including SLE. Reduced Treg numbers are observed in the peripheral blood of patients with SLE, particularly during active disease periods.55–57 The suppressive function of SLE-derived Tregs has also been studied, but the results are conflicting. Some reports claim they are unable to efficiently suppress proliferation and cytokine production.58,59 However, others suggest that the function of Tregs is conserved and that the suboptimal T-cell suppression observed in in vitro assays is the consequence of SLE T cells being abnormally resistant to Treg-induced suppression.60,61 One study has identified a CD8+ FoxP3+ regulatory cell subset present in patients with severe lupus subjected to autologous hematopoietic stem cell transplantation. Interestingly, the presence of these cells was associated with disease remission.62
Intrinsic T-Cell Defects
Assembly and Selection of the T-Cell Repertoire
Deficient removal of autoreactive T cells can cause autoimmunity. In patients with autoimmune polyendocrinopathy (APECED), absence of AIRE, a gene that allows the thymic expression of tissue-restricted antigens, hampers negative selection, allowing autoreactive T cells to egress the thymus.63 In patients with SLE, central tolerance has been studied indirectly through analysis of the frequency of peripheral blood autoreactive T cells.64,65 Histone-reactive T cells have been identified with a similar frequency in healthy controls, suggesting that negative selection occurs normally.64,65 In murine models of lupus, this process has also been evaluated on transgenic mice that express a specific preformed TCR.66,67 In mice with lupus-like diseases, thymocyte deletion is unremarkable when the cognate antigen is expressed in the thymus.66–68
Taken together, studies performed in patients and mice with lupus indicate that no gross defect in central tolerance processes underlies SLE. However, because T-cell selection is based on the molecules of the MHC, the array of MHC molecules present in each person determines the T-cell repertoire and the peptides to be presented in the thymus and during immune responses. Thus, even if no defects have been found in the central tolerance processes of patients with SLE, the characteristics of the T-cell repertoire created in the thymus are likely involved in the proclivity of patients with lupus to mount self-aimed responses. This likelihood may explain why the MHC locus is the region most commonly linked to lupus in genetic association studies.69
T-Cell Activation and Signaling
T-cell activation is abnormal in patients with SLE. Defects in key molecules involved in modulating the T-cell response to antigen presentation alter the signaling pathways elicited through the TCR. This phenomenon skews the expression of genes that control T-cell function.3,70
Intracellular residues of proteins associated to the TCR (CD3 complex) deliver activation signals into the cell by becoming phosphorylated following antigen recognition. The expression of a central component of CD3, the ζ chain, is decreased in T cells from patients with SLE.70 However, this decrease is paradoxically associated with an increased response to TCR stimulation. The reason is that CD3ζ is replaced by a closely related molecule, the common γ chain of the immunoglobulin receptor (FcRγ).71 The substitution of CD3ζ by FcRγ affects the intensity of the TCR-derived signal. FcRγ couples with spleen tyrosine kinase (Syk) instead of with ZAP-70 (ζ-associated protein 70).72 As a consequence, TCR engagement is followed by an abnormally high influx of calcium (Figure 9-3).73
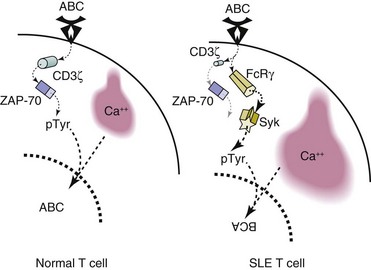
FIGURE 9-3 Structural differences alter the T-cell receptor (TCR) signaling process of the SLE T cell.
Decreased expression and altered membrane localization of CD3ζ is thought to be a central defect in this process. Interestingly, multiple molecular mechanisms have been described in SLE T cells that contribute to the diminished expression of CD3ζ. They include decreased production,74–76 decreased stability,77,78 and increased degradation.79
A closely related phenomenon described in SLE T cells is the presence of preaggregated lipid rafts in the T-cell membrane. These cholesterol-rich membrane areas carry signaling molecules and fuse at the pole of the cell where antigen is being presented. In quiescent T cells, lipid rafts are distributed throughout the cellular surface and coalesce after activation. The clustering allows signal transduction to occur effectively because all the necessary elements are rapidly drawn together. In T cells from patients with SLE, lipid rafts are clustered even in the absence of stimulation.80,42 This phenomenon likely contributes to the increased signal transduction observed after TCR stimulation in SLE T cells.81,82 Administration of a lipid raft–clustering agent accelerated disease onset in a murine model of lupus (MRL/lpr), whereas injection of a drug that disrupts lipid raft clustering had the opposite effect; these findings suggest that lipid raft clustering can indeed promote T-cell activation in vivo.83
The events that are initiated at the cell membrane when the TCR engages its cognate antigen are delivered through complex signaling pathways that cause immediate reactions in the cell and activate transcription factors. Activation of mitogen-activated protein (MAP) kinases mediates several cellular processes, such as proliferation, gene expression, and apoptosis induction. In T cells from patients with lupus, the MAP kinase activity is abnormal.84,85 The abnormality could contribute to autoimmunity, because MAP kinase function has been linked to maintenance of tolerance.86 In fact, mice deficient in RasGRP1 (RAS guanyl–releasing protein 1)87 or in protein kinase C (PKC) δ (a MAP kinase activator)88 demonstrate spontaneous autoimmune diseases.
Other signaling pathways are also affected in SLE T cells. Cyclic adenosine monophosphate (AMP)–dependent protein phosphorylation has been reported to be impaired,89 probably because of reduced levels of the protein kinase A.90 The activities of PKC and Lck (lymphocyte-specific protein tyrosine kinase) are also low in SLE T cells.91,92 In contrast, activity of the protein kinase PKR (involved in the phosphorylation of translation initiation factors) is increased.93 Likewise, activity of phosphatidylinositol-3 kinase (PI3K), the enzyme that produces the second messengers PIP2 and PIP3, is increased in mice with a lupus-like disease induced by alloreactivity.94 The importance of this pathway was further supported by studies proving that pharmacologic inhibition of class IB PI3K can ameliorate disease in MRL/lpr mice.95,96
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


