important for an orthopaedist to recognize this condition, since its identification allows for referral for management of the cardiovascular abnormalities, early treatment of which can prevent premature mortality.
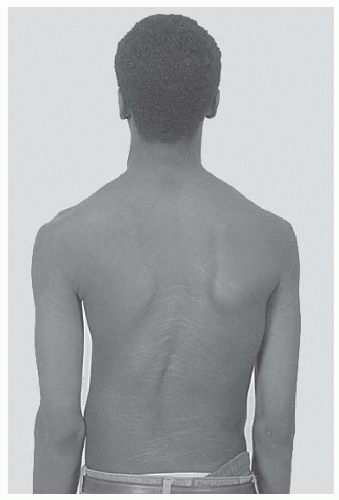 FIGURE 8-1. Stria in a boy with Marfan syndrome, who initially presented for evaluation of scoliosis. |
Although molecular diagnosis for a mutation in the fibrillin gene is available, this is usually not required in making the diagnosis, as physical findings and information from radiographic studies are generally sufficient for this purpose.
TABLE 8-1 Diagnostic Criteria for Marfan Syndrome: A Comparison of the Berlin land Ghent Diagnostic Criteria | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
reducing the amount of correction in a patient treated with a growing rod was shown in a case report to reverse cardiac failure (33). Computerized tomography (CT) scan to assess bony anatomy, especially of the pedicles, is quite useful in preoperative planning of hook and screw placement. Other unusual spinal deformities can occur, such as subluxation of vertebrae (25, 34). Traction should be used with caution, especially in cases with underlying kyphosis, as it can worsen and cause subluxation (26).
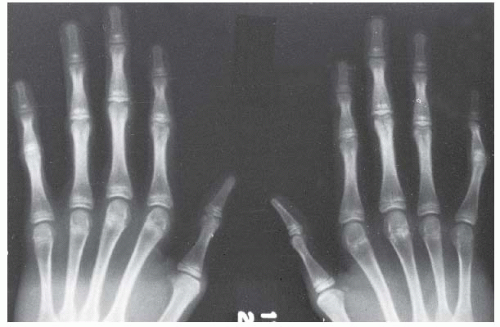 FIGURE 8-2. Hands showing arachnodactyly. Notice the long, thin metacarpals and phalanges. |
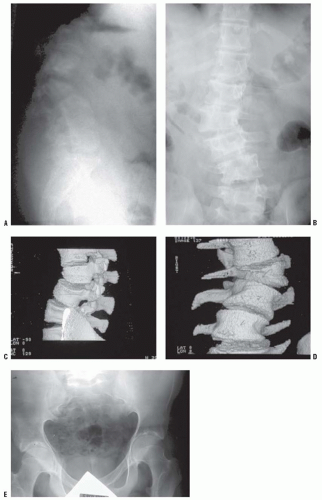 FIGURE 8-3. Scoliosis (A,B) and protrusia of the hips (E) in a patient with Marfan syndrome. C, D: Deformity of the apical vertebrae is shown in a three-dimensional reconstruction of a computerized tomographic scan image. (Courtesy of Chris Reily, MD, Vancouver, British Columbia, Canada.) |
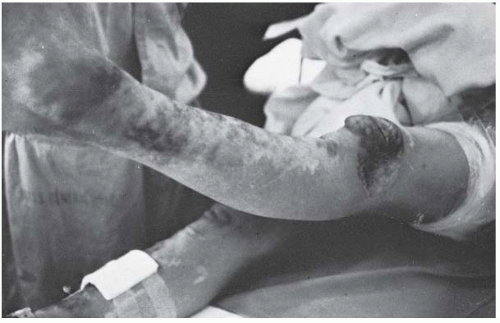 FIGURE 8-4. Patient with Ehlers-Danlos syndrome, type I. The knees and the pretibial regions have been subjected to recurrent injury and have accumulated heme pigmentation. (Courtesy of Michael G. Ehrlich, MD, Providence, Rhode Island.) |
physical therapeutic approaches, is often required. As opposed to individuals with normal joint laxity, patients with this condition have patellar instability in multiple planes (39). Since the matrix components that provide the mechanical properties to the soft tissues are defective, surgical approaches focusing on ligaments and tendons (e.g., soft-tissue procedures around the shoulder) have a low success rate. A variety of such operations are reported, such as osteotomies, which change the direction and location of insertion of tendons or osteotomies or that provide a larger joint area (tibial tubercle transfer operations for patellar dislocations, and femoral and pelvic osteotomies for hip subluxation). Procedures that involve surgery to the bones have a higher success rate than operations on ligaments or tendons. In particularly problematic cases, it may be necessary to place a bone graft to limit motion and prevent dislocation (e.g., a posteriorly placed graft at the elbow). Arthrodesis may be required as a last resort in those cases that remain symptomatic despite other managements (52—54).
TABLE 8-2 A Modified Classification Scheme for Ehlers-Danlos Syndrome | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
may be present at multiple levels, including nonadjacent sites (42). Valve problems can occur in EDS, so patients should have a cardiac evaluation before undergoing surgery. Low bone density is identified in EDS; however, when one corrects for the activity level of these patients, the bone density may not be so abnormal (56). Pharmacologic treatment for low bone density should be considered only in rare instances.
TABLE 8-3 Neurofibromatosis Type I: Diagnostic Criteria | ||||||||
|---|---|---|---|---|---|---|---|---|
|
similar to the coast of Maine. The spots vary greatly in number, shape, and size, and six lesions >1 cm in size are required for the diagnostic criteria. Axillary and inguinal freckling are common and serve as good diagnostic markers, because such freckling is exceptionally rare except in people with NF. Hyperpigmented nevi are dark brown areas that are sensitive to the touch; they typically overlie a deeper plexiform neurofibroma.
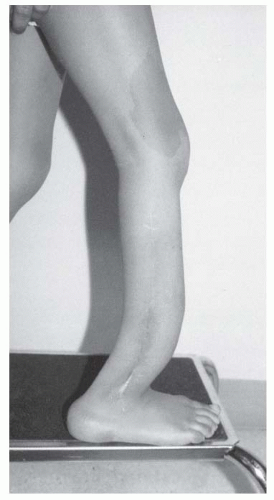 FIGURE 8-5. Neurofibromatosis in a 6-year-old child. Notice the large café-au-lait spot on the thigh and the anterior bowed tibia typical of pseudarthrosis. (From Goldberg MJ. The dysmorphic child: an orthopedic perspective. New York, NY: Raven Press, 1987, with permission.] |
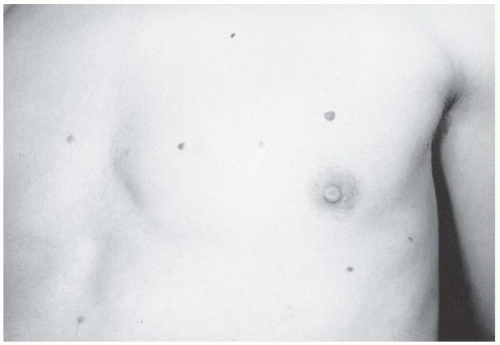 FIGURE 8-6. Neurofibromatosis in a 14-year-old patient. Cutaneous neurofibromas make their appearance with the onset of puberty. (From Goldberg MJ. The dysmorphic child: an orthopedic perspective. New York, NY: Raven Press, 1987, with permission.) |
diagnosis is established, further ophthalmologic evaluation is not necessary (53, 54).
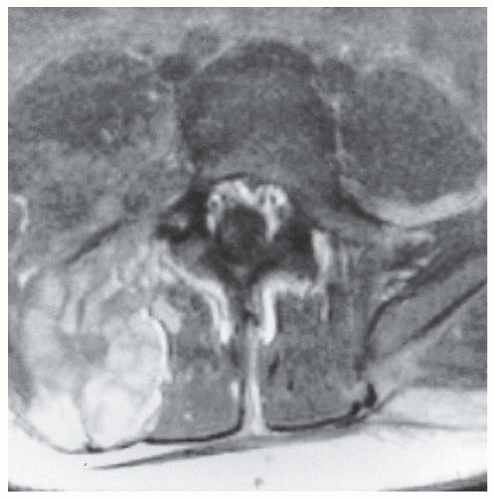 FIGURE 8-7. Neurofibromatosis in a 16-year-old patient. The MRI at the level of L4-L5 demonstrates a large plexiform neurofibroma that invades the neural axis. It extends from the level of L3 to the sacrum. |
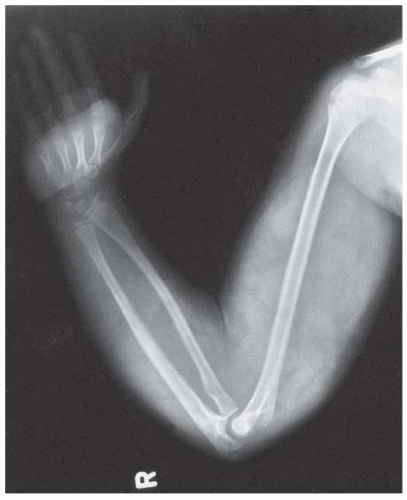 FIGURE 8-8. Neurofibromatosis in a 10-year-old patient. Hypertrophy affects the arm from the shoulder to the fingertips; the major component is soft tissue. Nodular densities throughout the upper arm are consistent with a plexiform neurofibroma. Notice the lack of skeletal overgrowth and some attenuation of the radius and ulna, caused by external compression by the neurofibroma. (From Goldberg MJ. The dysmorphic child: an orthopedic perspective. New York, NY: Raven Press, 1987, with permission.) |
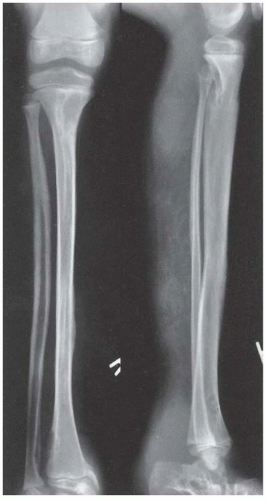 FIGURE 8-9. Neurofibromatosis in a 10-year-old patient. The radiograph shows an array of cystic and scalloped skeletal lesions in the tibia and os calcis of the right leg. Some of the lesions are characteristic of neurofibromatosis. Other lesions, occurring in isolation, can mimic benign fibrous tumors. Scalloped cortical erosion at the upper end of the femur, permeative bone destruction in the region of the os calcis, and metaphyseal cystic lesions are other features. (From Goldberg MJ. The dysmorphic child: an orthopedic perspective. New York, NY: Raven Press, 1987, with permission.) |
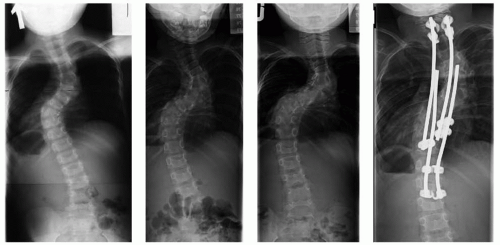 FIGURE 8-10. Neurofibromatosis in a 5-year-old patient. A dystrophic curve is shown in the left panel. There is a shortsegment scoliosis, with ribboned ribs show cystic irregularities. There was a delay in the recommendation for surgery, and the middle two panels show the rapid progression in the dystrophic curve over the next 12 months. The right panel shows the curve after undergoing surgery including anterior and posterior fusions of the dystrophic segments. |
severe cases, the spine can even seem to be “dislocated” because of the kyphosis and scoliosis. In cases with extremely severe deformity, halofemoral or halogravity traction may be necessary to safely straighten the spine to a more acceptable deformity without producing neurologic sequelae. Other reported techniques include inserting a bone graft without instrumentation and then gradually straightening the curve using a cast postoperatively (85). In rare severe cases in which there is a vertebral “dislocation,” one can use instrumentation to achieve an overall alignment of the back, while leaving the vertebrae “dislocated” (86). Unusual complications have been reported in the management of such dystrophic curves, such as a rib head migrating into the neural canal resulting in spinal cord compromise (87).
and distal synostosis to produce a single-bone forearm, the use of a vascularized fibula graft, or resection of the pseudarthrosis with shortening of the forearm and internal fixation (111). Pharmacologic approaches to the pseudarthrosis in NF are reported. A mouse model suggests the use of lovastatin, but the mouse does not develop pseudarthroses, only bowing of the bones, and as such human studies of this approach are needed (53). Direct installation of BMP to the pseudarthrosis site may help in the achievement of union, but variable results are reported, and it is not known if the use of BMP in patients with an inherited premalignant condition has long-term harmful consequences (80).
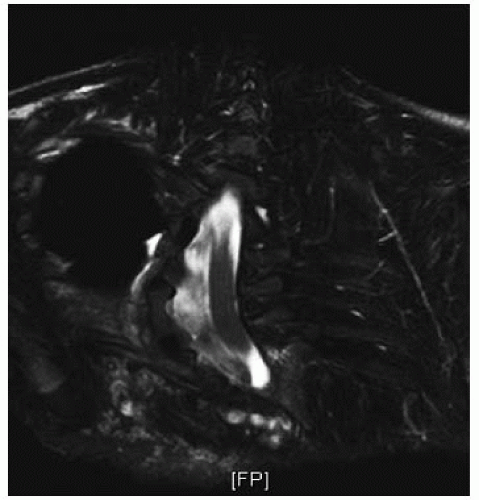 FIGURE 8-11. MRI of the spine of the patient shown in Figure 8-10, showing dural ectasia |
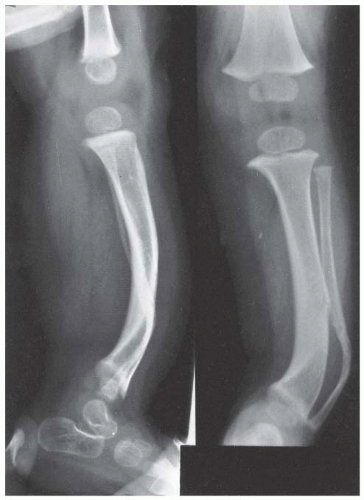 FIGURE 8-12. Neurofibromatosis in a 1-year-old patient. The anterolateral bow of the tibia and the fibula warrant concern about impending fracture and pseudarthrosis. (From Goldberg MJ. The dysmorphic child: an orthopedic perspective. New York, NY: Raven Press, 1987, with permission.] |
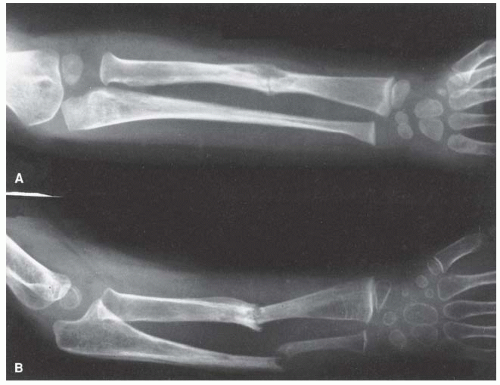 FIGURE 8-13. Neurofibromatosis in a 3-year-old patient. The radiograph shows progressive pseudarthrosis of the radius and ulna after a pathologic fracture. A: Fracture through the cystic lesion of the radius and thinning of the midulna. B: After 10 months of cast immobilization, pseudarthrosis affects the radius and ulna. (From Goldberg MJ. The dysmorphic child: an orthopedic perspective. New York, NY: Raven Press, 1987, with permission.) |
accounts for the large organs: in the adrenals, giant cortical cells; in the gonads, an increased number of interstitial cells; and in the pancreas, islet cell hyperplasia. This underlies the 10% risk of developing benign or malignant tumors. Wilms tumor is the most common.
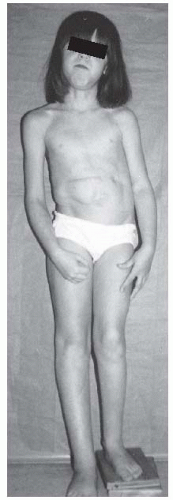 FIGURE 8-14. Beckwith-Wiedemann syndrome in an 8-year-old patient. Hemihypertrophy on the right, a part of this syndrome, is combined with hemiatrophy on the left, caused by acquired encephalopathy secondary to hypoglycemic seizures as a newborn, leading to a significant leg-length discrepancy of 4.6 cm. Abdominal scars are a consequence of omphalocele repair. (From Goldberg MJ. The dysmorphic child: an orthopedic perspective. New York, NY: Raven Press, 1987, with permission.) |
averages approximately 2 cm at maturity, but can be as much as 6 cm. Regardless of the magnitude of the discrepancy, it is clinically more apparent because the child is small. The face is characteristically triangular and seemingly too small for the cranial vault. There have been several reports of variations in sexual maturation pattern and malformations of the genitourinary system.
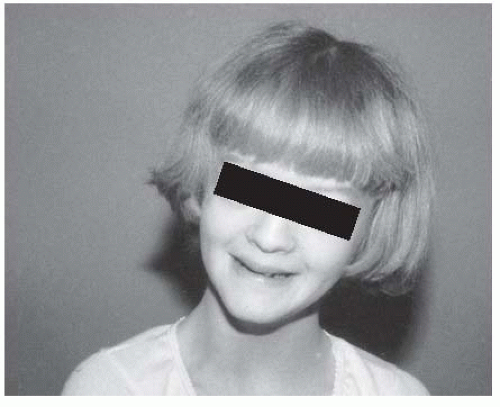 FIGURE 8-15. Russell-Silver syndrome. The triangular face is seemingly small for the size of the skull. |
finger, followed by the long finger and the thumb. It is the second toe that is most commonly macrodactylous. The regional sensory nerve is greatly increased in size, taking a tortuous route through the fatty tissue.
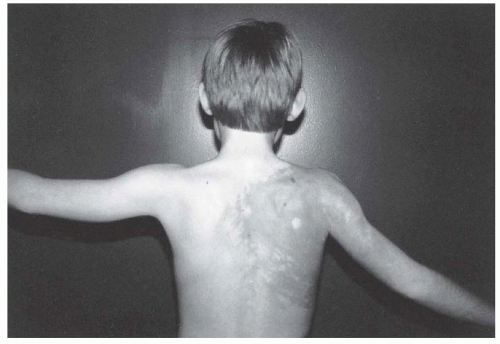 FIGURE 8-16. Proteus syndrome. Notice the cutaneous markings, large hemangioma of the shoulder, and lightly pigmented area on the back. There is some atrophy of the shoulder and arm muscles and a fixed contracture of the elbow. |
in a musculoskeletal malformation. These disorders can be identified at birth, because the problem is present at the start of development. Despite this, sometimes, the abnormalities do not become obvious to parents or physicians until the child is older. Because these are generally patterning problems, surgery to correct malalignment is usually quite successful. There are frequently manifestations in other organ systems, because the same developmental signaling pathways play important roles in the development of multiple organs. These disorders are not associated with an increased rate of neoplasia. Symptoms from the malformations often increase with age because the abnormally shaped structures cannot sustain the stresses of normal activity. This results in the early development of degenerative problems. These disorders are usually inherited in an autosomal dominant manner, although the inheritance pattern is more variable than in disorders caused by genes encoding for structural proteins or for proteins implicated in neoplasia.
severity of the anomalies and they are frequently associated with other malformations (176, 177). It has an estimated incidence of 1 in 5600 births (178), and roughly 2% of individuals with congenital spinal abnormalities will have another manifestations of ocular—auricular—vertebral dysplasia (138).
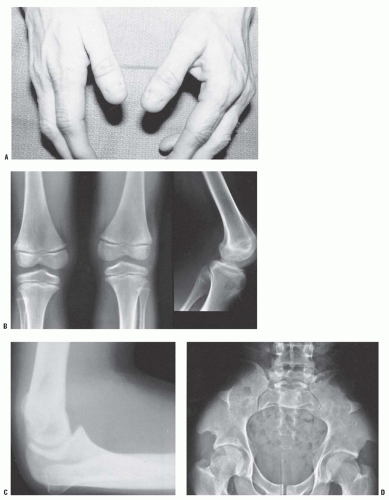 FIGURE 8-17. Nail-patella syndrome. The classic quartet of features consists of dystrophic nails (A), absent patellae (notice the region of osteochondritis dissecans on the lateral film) (B), posterior dislocation of the radial head (C), and iliac horns (D). |
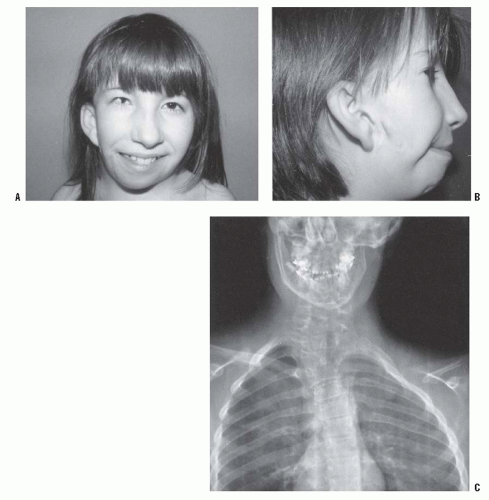 FIGURE 8-18. Goldenhar syndrome. A: Facial asymmetry and epibulbar dermoid of the right eye. B: Malformed ears with preauricular tags and sinuses. C: X-ray film demonstrates the congenital anomalies of the lower cervical and the upper thoracic spine. Hypoplasia of the ascending ramus of the mandible accounts for the facial asymmetry. The clavicle is absent on the same side as the deformation of the face. (From Goldberg MJ. The dysmorphic child: an orthopedic perspective. New York, NY: Raven Press, 1987, with permission.) |
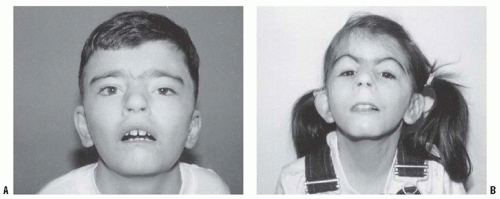 FIGURE 8-19. Cornelia de Lange syndrome. Notice the classic facial features of heavy eyebrows meeting in the midline, upturned nose, downturned corners of the mouth, and long eyelashes in a 13-year-old boy (A) and a 7-year-old girl (B). (From Goldberg MJ. The dysmorphic child: an orthopedic perspective. New York, NY: Raven Press, 1987, with permission.) |
Almost all of them walk, but their milestones are delayed. There is retarded mentation, but the added features of no speech and no interactions cause major disability (201). Self-mutilating behavior can be an obstacle to orthopaedic care (202, 203).
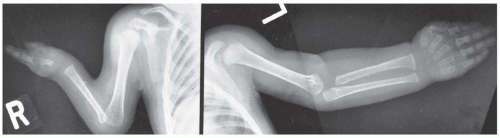 FIGURE 8-20. Cornelia de Lange syndrome: a child with a severely affected upper extremity on her right side (i.e., absent ulna and fingers) and a mildly affected arm on her left (i.e., short thumb and dysplasia of proximal radius). (From Goldberg MJ. The dysmorphic child: an orthopedic perspective. New York, NY: Raven Press, 1987, with permission.) |
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


