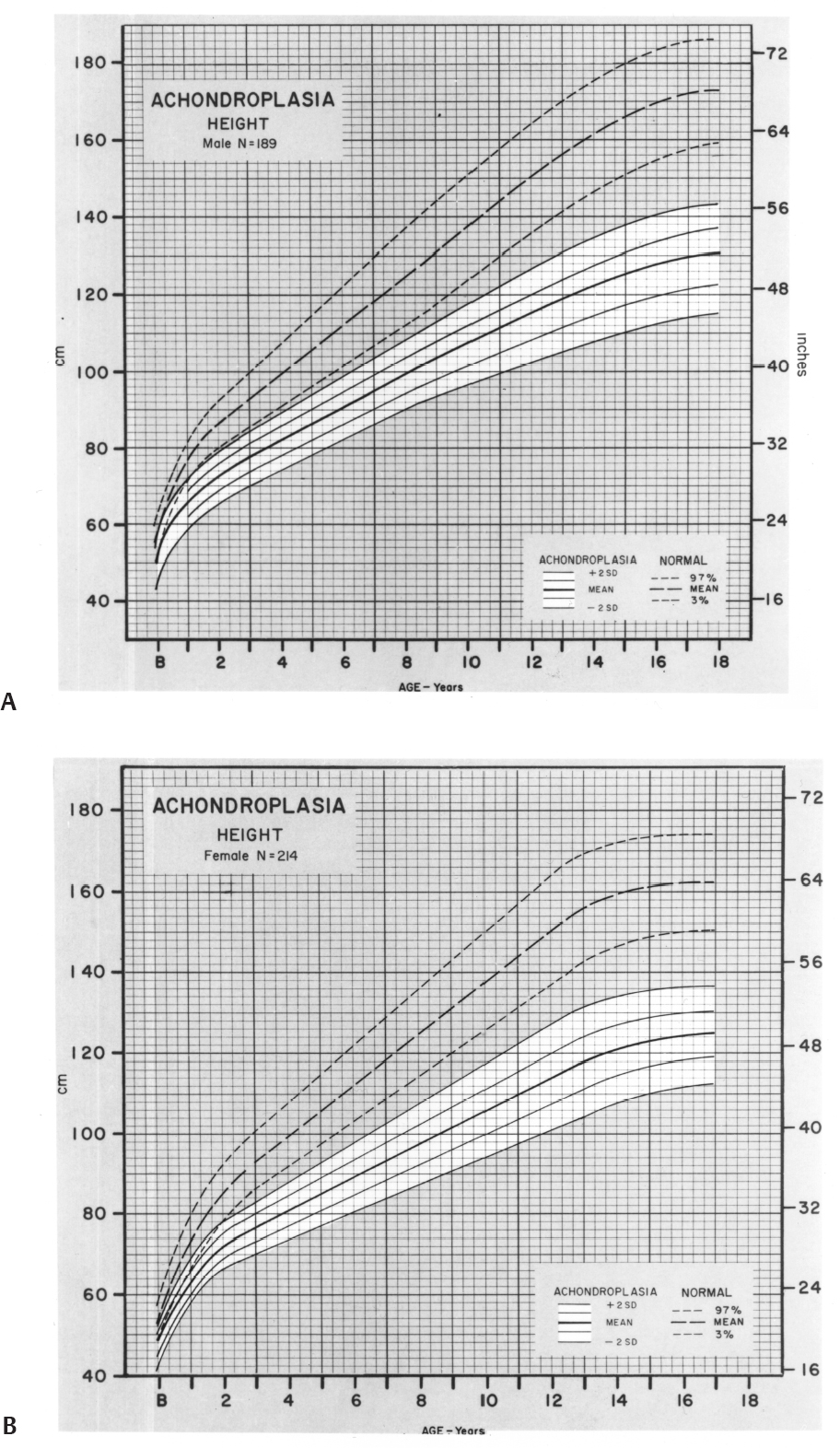
3 Skeletal Syndromes and Systemic Disorders in Pediatric Orthopedics This chapter summarizes developmental syndromes that involve skeletal abnormalities. A diagnosis or discussion of prognosis may be required. Because knowledge of a large number of facts rather than basic principles is required, this chapter was created to help. It is organized according to presenting features (see below). More detailed discussion of these conditions is available in the cited references. Skeletal dysplasis involve basic and systemic abnormalities of bone and cartilage growth and development. Usually short stature occurs. 1. Achondroplasia a. Autosomal dominant with frequent new mutations. Most common skeletal dysplasia. b. Genetic defect: Fibroblast growth factor receptor protein 3 (a gain-of-function mutation). c. Major features 1) Midface hypoplasia 2) Rhizomelic dwarfism 3) Genu varum (variable) 4) A 3- to 6-month delay in motor milestones 5) Thoracolumbar kyphosis, often resolving with growth 6) Spinal stenosis is greatest in the lumbar spine, greatest distally, but may affect entire spine, including foramen magnum, and cause severe developmental delay. d. Height graph (Fig. 3.1) e. Treatment: Monitor for spinal stenosis and persistent kyphosis. Correction of knee and ankle deformities at patient’s discretion. Limb lengthening is an option and is usually successful but time consuming. 2. Pseudoachondroplasia a. Although these patients are also rhizomelic, the epiphyseal involvement in this syndrome causes major differences from achondroplasia. b. Genetic defect: COMP (cartilage oligomeric matrix protein); found in extraterritorial matrix). Fig. 3.1 Comparison of growth patterns of normal-stature and achondroplastic persons. (A) Males. (B) Females. (From Horton WA, Rotter JI, Rimoin DL, Scott CI, Hall JG. Standard growth curves for achondroplasia. J Pediatr. 1978;93(3):436 (Figs. 1 and 2). Reprinted with permission.) 1) Rhizomelic shortening of extremities 2) Variable knee deformities (often varus on one side, valgus on the other) 3) Mild platyspondyly; minimal scoliosis; no stenosis 4) Odontoid hypoplasia, possible C1-C2 instability 5) Epiphyseal deformation; eventual degeneration 6) Ligamentous laxity d. Treatment: Screen cervical spine; correct major limb malalignment. Many patients require joint arthroplasty as adults. 3. Diastrophic dysplasia a. Autosomal recessive b. Genetic defect: Diastrophic sulfate transporter (DTST) c. Major features 1) “Cauliflower ear” developing at around 6 months of age 2) Rhizomelic shortening of extremities 3) Contractures of major joints with later degenerative joint disease (DJD) 4) Hands: Hitchhiker thumb, symphalangism 5) Dislocated hips: Occasionally 6) Equinovarus or other foot deformities 7) Cervical spina bifida with severe kyphosis: Sometimes resolves 8) Scoliosis of thoracic and lumbar spine d. Treatment: Screen and monitor the cervical spine. Correct foot deformities, scoliosis, and limb contractures. Arthroplasty as indicated. 4. Spondyloepiphyseal dysplasia congenita a. Autosomal dominant with frequent new mutations b. Genetic defect: Collagen 2A1 c. Major features 1) Extreme short stature 2) Odontoid hypoplasia/os odontoideum: May have instability 3) Platyspondyly, scoliosis 4) Coxa vara, epiphyseal irregularity, DJD d. Treatment: Screen or stabilize the cervical spine. Correct scoliosis as indicated. Treatment for hip dysplasia is of uncertain benefit. Joint arthroplasty is often indicated in adulthood. 5. Spondyloepiphyseal dysplasia (SED) tarda a. Variable transmission; diagnosis late in childhood b. Genetic defect: SEDL (a tracking protein) or others c. Major features 1) Irregular ossification, DJD 2) Hips may resemble Perthes, but in SED bilaterally synchronous 3) Osteoarthritis of other joints 4) Scoliosis 6. Multiple epiphyseal dysplasia a. Autosomal dominant b. Gene defect: COMP (found in matrix) or collagen 9 or DTST c. Major features 1) Variable; usually mild short stature because of short limbs 2) Irregular epiphyseal ossification with deformity, pain, DJD 3) Hips, knees, ankles are most involved. Patella may show “double layer.” 4) Usually presents in late childhood to adulthood 7. Metatropic dysplasia a. Major features 1) Epiphyseal or metaphyseal enlargement: “Knobby” joints with contractures 2) Cervical stenosis, instability 3) Scoliosis, kyphosis, later onset 4) Coccygeal tail 5) Thoracic hypoplasia; may cause respiratory compromise 6) Initially short-limb dwarfism; becomes short-trunk type with onset of scoliosis 8. Chondrodysplasia punctata (Conradi–Hunerman) a. Autosomal dominant, recessive, and X-linked b. Major features 1) Multiple asymmetric epiphyseal calcifications 2) Rhizomelic form may have cervical stenosis or kyphosis as well as thoracolumbar scoliosis. 3) Good prognosis for dominant form. Decreased life expectancy for other forms 9. Multiple hereditary exostoses (MHE) a. Inheritance: Autosomal dominant b. Genetic defect: At least three have been described; EXT-1 and -2 on different chromosomes. EXT-1 produces more serious form. c. Clinical appearance: Mild short stature d. Categories of problems 1) Local impingement on tendons, nerves, spinal canal, ribs 2) Asymmetrical growth in two-bone segments (forearms and legs) leading to valgus at knees, ankles, elbow, and wrists and possibly radial head dislocation. 3) Leg-length inequality (usually less than 4 cm) 4) Risk of malignant degeneration (in about 1% of patients) 5) Osteochondromas may grow silently in spinal canal; monitor neurologic examination. 6) Patients with MHE often heal incisions with wide scars or keloids. e. Radiographic features 1) Osteochondromas in metaphysis, pointing away from joint 2) Cortex of osteochondroma is confluent with that of host bone. 3) May be sessile or pedunculated f. Treatment 1) Resect lesions only when symptomatic. Increased rotation not predictable in forearm. 2) Correct knee and ankle valgus when greater than 10 degrees. 3) Monitor in adulthood every 2 years, possibly with bone scan. 4) Obtain spine magnetic resonance imaging (MRI) if there is any question of involvement. 10. Dysplasia epiphysialis hemimelica (Trevor disease) a. Definition: Epiphyseal osteochondroma; no genetic pattern b. Clinical features: Presents in first decade of life; restricted joint motion, enlarged joint, or locking. Knee, foot, and ankle are most commonly involved. c. Radiographs: Multiple opacities in exostotic cartilage; these eventually coalesce. d. Treatment: Resection, attempting to preserve normal cartilage Kettelkamp DB, Campbell CJ, Bonfiglio M. Dysplasia epiphysealis hemimelica: a report of fifteen cases and a review of the literature. J Bone Joint Surg Am. 1966;48(4):746–765, discussion 765–766 11. Multiple enchondromas (Ollier disease) a. Genetic defect: PTH/PTHRP b. Clinical presentation 1) Angular deformity 2) Bony irregularity 3) Limb-length inequality c. Radiographic features 1) Diffuse enchondromas in metaphysis; occasionally epiphyses. Usually asymmetrical d. Treatment 1) Angular or length correction of limb 2) Monitor for malignancy, especially in Maffucci syndrome 12. Cleidocranial dysplasia a. Autosomal dominant b. Genetic defect: CBFA1, a transcription factor c. Clinical features 1) Persistently open skull sutures with bulging calvarium 2) Hypoplasia or aplasia of clavicles 3) Wide symphysis pubis 4) Hip abnormalities (coxa vara) 5) Short middle phalanx of fifth finger 6) Scoliosis with or without syringomyelia 7) Multiple dental abnormalities Jensen BL. Somatic development in cleidocranial dysplasia. Am J Med Genet. 1990; 35(1):69–74 13. Dyschondrosteosis (Leri–Weill disorder) a. Genetic defect: SHOX pseudoautosomal genes b. Major features 1) Mild short stature (<25 percentile) 2) Madelung deformity (dorsoulnar deficiency of distal radial growth) 3) Relative shortening of forearm and leg; varus or valgus deformity 4) Females predominant 1. Cornelia de Lange (Brachmann de Lange type) a. Genetic defect: NIPBL; or microdeletion on chromosome 3 b. Major features 1) Synophrys (single eyebrow) 2) Down-turned mouth 3) Mandibular spur in infancy 4) Hirsutism 5) Gastroesophageal reflux 6) Small for gestational age, with continued growth retardation 7) Motor, speech, and intellectual delay 8) Cardiac abnormalities c. Orthopedic involvement 1) Upper extremity anomalies (~100%) a) Micromelia, phocomelia b) Decreased number of fingers c) Lobster-claw hand d) Proximally placed thumb e) Elbow anomalies 2) Lower extremities a) Miscellaneous foot deformities and contracture b) Avascular necrosis (AVN) of femoral head in 10% d. Treatment: Correct lower extremity abnormalities if limiting ambulation; upper extremities: individualized treatment 2. Riley–Day familial dysautonomia a. Ashkenazi Jews only: Autosomal recessive b. Sympathetic overactivity is key feature c. Major features 1) Deficient sensation of pain and proprioception 2) Gastroesophageal reflux, pneumonia 3) Variable life expectancy d. Orthopedic abnormalities and implications 1) Scoliosis/kyphosis before age 8; poor brace tolerance; fuse early 2) Fractures from osteopenia or dyscoordination 3) AVN of femoral head, distal femur, talus 4) Hip dysplasia a. Autosomal dominant, normal life expectancy b. Orthopedic features 1) Nails grooved, small, or absent, especially on thumb 2) Multiple knee anomalies: patella tripartite, small or absent, lateral femoral condyle hypoplastic, (valgus) osteochondritis disse-cans of lateral femur and talus 3) Elbow: Capitellar hypoplasia, cubitus valgus, flexion contracture 4) Iliac horns 5) X-linked hypophosphatemic rickets 1. Fibrodysplasia ossificans progressiva a. Progressive, disabling heterotopic ossification or ankylosis. Incidence 1:1,000,000 b. Etiology: Enhanced signaling of BMP4 c. Characteristic shortening/valgus of great toe d. Ossification starts as tender, hard nodule; progresses proximal to distal, posterior to anterior e. Do not biopsy; may accelerate the process f. Genetics: Usually a spontaneous mutation but may be transmitted as autosomal dominant 2. Progressive diaphyseal dysplasia (Camurati–Engelman disease): a. Clinical features: Pain, fatigue, muscle atrophy b. Etiology: activating mutation in transforming growth factor beta (TGF-β) genes c. Radiographs: Symmetrically widened, sclerotic diaphyses; epiphyses spared tibia, femur most commonly involved c. Treatment: Osteotomies only if marked deformity. Possible role for bisphosphonates 3. Melorheostosis a. Syndrome involving asymmetrical extraosseous longitudinal hyperostotic streaks resembling molten wax; limb pain and soft tissue contracture b. Treatment: Analgesics, bracing contracture releases, and bone shortening a. Linear intraosseous metaphyseal striations b. Autosomal dominant c. Asymptomatic d. No treatment required 5. Osteopoikilosis a. Multiple symmetrical intraosseous epiphyseal–metaphyseal “spots” b. Autosomal dominant c. Asymptomatic 1. Background: Inheritance—somatic mutation that produces mosaic distribution of lesions in one (monostotic) or many (polyostotic) bones a. The molecular basis is a postzygotic activating mutation in the GNAS1 gene, which encodes for the cyclic adenosine monophosphatase (cAMP)-regulating α subunit of the Gs protein complex. b. Histologically the fibrous tissue undergoes ossification to small, irregular trabeculae (“alphabet soup”). The disease process is most active during growth and causes weakening of the bone and pathologic fracture. 2. Clinical manifestations: Monostotic accounts for 80% of cases. Polyostotic is found in several bones on one side of the skeleton or scattered throughout the skeleton. In the axial skeleton, the craniofacial bones and the ribs are the most common sites; in the appendicular skeleton, the tibia and proximal femur are the most common sites. a. Monostotic fibrous dysplasia usually presents without symptoms, and the lesion is found when a radiograph is taken for unrelated reasons. b. Polyostotic fibrous dysplasia often results in distortion of the skeletal and facial configuration. The peak incidence of fractures is during the first decade of life, followed by a decrease thereafter. Lesions of the femoral neck may cause progressive coxa vara, leading to the shepherd’s crook deformity; this is the most common angular deformity in polyostotic fibrous dysplasia. Spinal involvement and scoliosis may also occur. c. Skin lesions include café-au-lait spots with an irregular border (“coast of Maine”). 3. Imaging findings: Elongated lesion with symmetric cortical thinning and outward expansion, the characteristic diaphyseal “long lesion in a long bone.” Lesion shows few trabecular markings and has a ground-glass appearance. Some may be entirely radiolytic or radiodense. There may be an associated angular deformity. Fibrous dysplasia shows excessive uptake on bone scan. 4. Treatment: Unnecessary for asymptomatic lesions of fibrous dysplasia. Large or symptomatic lesions may be treated by curettage and allografting. Lesions of the femoral neck should be treated with metallic support with or without cortical bone grafting because of the risk of fatigue fracture. If symptomatic varus deformity is present, treatment should include valgus osteotomy with cortical bone grafting and rigid internal fixation. Because of poor bone quality, intramedullary fixation is preferable to plates and screws alone for lesions of the femoral shaft. Deformity may occur at stress risers, similar to osteogenesis imperfecta. Medical management includes the use of bisphosphonates. Early studies suggest that bisphosphonates decrease pain, improve the radiologic appearance of the lesions, and decrease the fracture rate. 1. Marfan syndrome (MFS) a. This is a disorder of fibrillin-1, which also affects TGF-β distribution and has multiple effects on the skeleton and connective tissue. Because some features may be seen in the general population, the following diagnostic criteria have been developed. b. Diagnostic (Ghent) criteria: Two major criteria (*below) and involvement of another system; system involvement (*indicates major manifestation) 1)
 Skeletal Dysplasias
Skeletal Dysplasias
Bibliography
Bibliography
 Other Syndromes Involving Short Stature
Other Syndromes Involving Short Stature
 Sclerosing Bone Disorders
Sclerosing Bone Disorders
 Fibrous Dysplasia
Fibrous Dysplasia
 Marfan and Related Disorders
Marfan and Related Disorders
![]()
Stay updated, free articles. Join our Telegram channel


Full access? Get Clinical Tree


