Chapter 20 Role of Complement in Systemic Lupus Erythematosus Pathogenesis
INTRODUCTION
The complement system is intimately linked to SLE. Although paradoxical, this association has been recognized for decades. Two early observations formed the foundation for this conundrum. Vaughan and colleagues first assayed serum complement in four cases of SLE in 1951, and determined that diminished CH50 correlated with disease activity.1 This was confirmed by Elliott and Mathieson, who noted that complement depression was particularly associated with “albuminuria.”2 Lange and colleagues discovered that although low complement was characteristic of SLE whether or not there was renal involvement3 it was diminished in virtually all cases of acute, but not chronic, glomerulonephritis. Schur and Sandson found CH50 to be below 50% of normal in 46% of patients with renal involvement and 24% of patients with active SLE, but in only 4% of those with inactive disease—suggesting that complement levels were of particular value in following and evaluating patients with active SLE, especially those with nephritis.4 Thereafter, a large body of work demonstrated that complement activation (reflected by diminished serum levels of C3, C4, and CH50) plays a major role in the tissue inflammation and organ damage that result from lupus pathogenesis.
Several other seemingly contradictory observations demonstrated a strong association between hereditary homozygous deficiency of the classical pathway components and development of SLE.5–7 In fact, inherited complement deficiency is recognized as conferring the greatest known risk for development of SLE. Thus, paradoxically, complement deficiency may be causative in SLE. However, activation of this same inflammatory cascade is detrimental in patients who already have the disease.
COMPLEMENT BIOLOGY
The complement system is composed of more than 30 plasma and membrane-bound proteins that form three distinct pathways and that protect against invading pathogens: classical, lectin, and alternative8–11 (Fig. 20.1). Functionally inactive complement pro-proteins exist in plasma until their activation is triggered. Once activated, the proteins undergo a cascade of sequential serine protease—mediated cleavage events within each pathway, thereby releasing biologically active fragments and self-assembling into multimolecular complexes. In general, activation of the complement system can be viewed as a two-stage process. In the first stage, unique to each of the three activation pathways, the early complement components are activated and lead to the formation of the C3 convertases. In the second stage, all three pathways converge to form a lytic complex, which consists of the terminal complement components (Fig. 20.1).
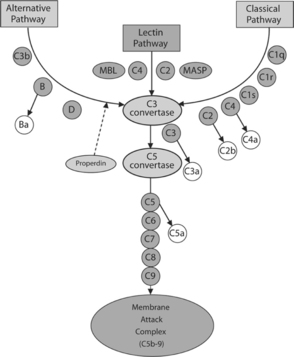
Complement Activation Pathways
The classical pathway of complement activation plays an important role in SLE pathogenesis and is responsible for executing a major effector mechanism of antibody-mediated immune responses. Five proteins are specific to the activation of the classical pathway: C1q, C1r, C1s, C4, and C2 (Fig 20.1). C1q sets into motion a sequential cascade of events when it binds to the Fc portion of IgG (particularly IgG1 and IgG3) or IgM molecules that are bound to an antigen. This process then activates C1r (a serine protease), which in turn leads to activation of C1s (also a serine protease). When C1s enzymatically cleaves the other two classical pathway proteins, C4 and C2, two small soluble polypeptides (C4a and C2b) are generated and released. Concurrently, a surface-bound bimolecular complex (C4b2a, which functions as an enzyme and is referred to as the classical pathway C3 convertase) is formed. A variety of other molecules can also initiate activation of the classical pathway by interacting with C1q. These include C-reactive protein,12 amyloid P component,13 β-amyloid protein,14,15 and DNA.16
The lectin pathway shares several components with the classical pathway (Fig. 20.1). It is initiated when mannose-binding lectin [MBL; also known as mannose-binding protein (MBP)] binds to a variety of repetitive carbohydrate moieties such as mannose, N-acetyl-D-glucosamine, and N-acetyl-mannosamine, which are abundantly present on a variety of microorganisms.17,18 MBL, a plasma protein composed of a collagen-like region and a carbohydrate-binding domain, is structurally similar to C1q. MBL forms complexes in the plasma with other proteins such as mannose-binding protein-associated serine proteases (MASPs).19–21 Under physiologic conditions, MBL does not bind to mammalian cells, probably because these cells lack mannose residues on their surface. Once bound to microbial pathogens, MASPs can cleave C4 and initiate the complement cascade. Alternatively, MBL (in place of C1q) may trigger the activation cascade by activating C1r and C1s. At this point, the lectin pathway intersects with the classical pathway and a C3 convertase (i.e., the C4b2a complex) is eventually generated.
In contrast to the classical and lectin pathways, activation of the alternative pathway is not dependent on antibodies or other specific recognition molecules. Three plasma proteins (Factor B, Factor D, and properdin) are unique to the alternative pathway (Fig. 20.1). Normally, native C3 molecules undergo a so-called “C3 tickover” process—a continuous low-rate hydrolysis of the thioester group that generates iC3* (hydrolyzed C3) and subsequently C3b fragments.22 Via the formation of thioester bonds, some of these spontaneously generated C3b fragments may covalently attach to the surface of microbial pathogens and host cells.
Effector Functions of Complement
The complement system protects against invasion by four primary and basic biological functions: opsonization, activation of inflammation, clearance of immune complexes, and osmotic lysis of invading microorganisms.8–11 During SLE pathogenesis, self-antigens and autoimmune complexes (rather than foreign microbes) generate complement activation and its inflammatory consequences.
C3a, C4a, and C5a are soluble proteolytic fragments, which are highly potent and pro-inflammatory. By binding to specific receptors expressed on leukocytes (e.g., C5a receptor), they attract and activate them. The larger fragments (C3b, C4b) and their derivatives (e.g., iC3b and iC4b) can remain bound to the surface of microbial pathogens (or autoantigens) and facilitate recognition and uptake of opsonized particles by phagocytic cells via specific receptors (e.g., complement receptors 3 and 4, CR3 and CR4) expressed on the latter.
When C3b and C4b bind to immune complexes, this binding prevents their aggregation into insoluble complexes and enhances their clearance. To mediate the clearance of C3b/C4b-opsonized immune complexes, erythrocytes that express CR1 transport these complexes to macrophages of the reticuloendothelial system in the spleen and liver.23,24 In addition, these complexes may bind to monocytes and neutrophils, which phagocytose them (often releasing lysosomal enzymes in the process). Finally, the C5b-9 MACs may perturb the osmotic equilibrium or disrupt the integrity of the surface membrane of target cells, thereby causing lysis of these cells.
For several decades, the role of the complement system was thought to be limited to these four effector functions. Recently, however, many additional roles for complement (such as a link between innate and adaptive immunity) have been discovered.25,26 These novel functions are discussed in the material following.
Regulators of Complement Activation and Complement Receptors
In humans and other mammals, a redundant family of regulatory proteins controls complement activation to ensure that host cells and tissues do not inappropriately activate this effective machinery (Table 20.1). Soluble or cell-surface regulatory molecules act at multiple steps of the activation pathways to control the potent consequences of complement activation. To do so, they use different mechanisms, functioning as proteolytic enzymes, cofactors for proteolytic enzymes, or competitive inhibitors of multimolecular convertases (Fig. 20.2).
TABLE 20.1 COMPONENTS OF THE HUMAN COMPLEMENT SYSTEM
| Effector Protein | Mr (kD) | Function/Pathway Involved |
|---|---|---|
| C1q | 450 (six-subunit bundle) | Recognition, binding/classical |
| C1r | 85 | Serine protease/classical |
| C1s | 85 | Serine protease/classical |
| C4a | 205 (3-chain, αβγ, complex) | Serine protease (C4b); anaphylatoxin (C4a)/classical |
| C2 | 102 | Serine protease (C2a); small fragment with kinin-activity (C2b)/classical |
| C3b | 190 (2-chain, αβ, complex) | Membrane binding, opsonization (C3b); anaphylatoxin (C3a)/terminal |
| C5 | 190 (2-chain, αβ, complex) | MAC component (C5b), anaphylatoxin (C5a)/terminal |
| C6 | 110 | MAC component/terminal |
| C7 | 100 | MAC component/terminal |
| C8 | 150 (3-chain, αβγ, complex) | MAC component/terminal |
| C9 | 70 | MAC component/terminal |
| Factor B | 90 | Serine protease/alternative |
| Factor D | 24 | Serine protease/alternative |
| Properdin | 55 (monomers); 110, 165, 220, or higher (oligomers) | Stabilizing C3bBb Complexes/alternative |
| MBL | 200-400 (2-4 subunits with three 32 KD chains each) | Recognition, binding/Lectin |
| MASP-1 | 100 | Serine protease/Lectin |
| MASP-2 | 76 | Serine protease/Lectin |
| Membrane-Bound Regulatory Protein | Mr (kD) | Function |
| CD35 (CR1c) | 160-250 (4 isoforms) | Binding C3b and C4b; cofactor for Factor I |
| CD46 (MCPd) | 45-70 (different glycosylation forms) | Promoting C3b and C4b inactivation by Factor I |
| CD55 (DAFe) | 70 | Accelerating decay of the C3bBb and C4b2a complexes |
| CD59 (protectin; H19) | 18-20 | Preventing C9 incorporation into the MAC in a homologous restriction manner |
| Soluble Regulatory Protein | Mr (kD) | Function |
| C1-inhibitor (C1-INH) | 105 | Removing activated C1r and C1s from the C1 complex |
| C4-binding protein (C4bp) | 570 (7-subunit complex) | Displacing C2b in the C4bC2b complex; cofactor for Factor I |
| Factor H | 160 | Displacing Bb in the C3bBb complex; cofactor for Factor I |
| Factor I | 88 | Serine protease cleaving C3b and C4b |
| Clusterin | 70 | Preventing insertion of soluble C5b-7 complexes into cell membranes |
| S protein (vitronectin) | 84 | Preventing insertion of soluble C5b-7 complexes into cell membranes |
| Carboxypeptidase N | 280 (multi-subunit complex) | Inactivating anaphylatoxins |
| Complement Receptor | Structure/Mr (kD) | Complement Ligand(s)f |
| CR1 (CD35) | Single chain, 190-280g | C3b, C4b, iC3b, C1q |
| CR2 (CD21) | Single chain, 140-145 | C3dg/C3d, iC3b |
| CR3 (CD11b/CD18) | 2-chain, α/β; 170/95 | iC3b |
| CR4 (CD11c/CD18) | 2-chain, α/β; 150/95 | iC3b |
| cC1qR (calreticulin) | Single chain, 60 | C1q (collagenous tail), MBL |
| gC1qR | Tetramer, 33/subunit | C1q (globular head) |
| C1qRP | Single chain, 126 | C1q (collagenous tail) |
| C3a receptor | Single chain, 50? | C3a |
| C5a receptor (CD88) | Single chain, 50 | C5a |
a. Serum concentration range considered normal: 20-50 mg/dL.
b. Serum concentration range considered normal: 55-120 mg/dL.
f. Noncomplement ligands (e.g., Epstin-Barr virus for CR2 and fibrinogen for CR3 and CR4) not listed.
g. Four allotypes with different numbers of SCR and displaying distinct Mr under reducing condition: CR1-A (220 kD), CR1-B (250 kD), CR1-C (190 kD), and CR1-D (280 kD).
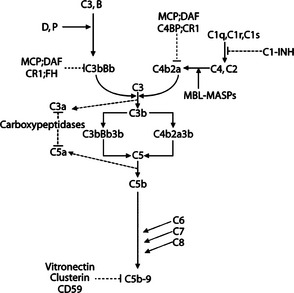
Receptors for proteolytic fragments of complement proteins (such as C3b, C4b, iC3b, and C3d or C1q) are expressed by a wide spectrum of cells and serve pivotal roles in executing many of the effector functions of complement described previously. Recent research has led to the identification of at least four receptors for C1q: cC1qR (calreticulin, a collectin receptor), gC1qR, C1qRp, and CR1 (CD35).27–30 C1q receptors bind opsonized immune complexes to endothelial cells. This induces expression of adhesion molecules on these cells and enhances leukocyte binding/extravasation.31 On other cell types, C1q binding leads to enhanced phagocytosis, increased generation of reactive oxygen intermediates, and activation of platelets, presumably via distinct receptors.32–34
Two major receptors for C3- and C4-derived fragments, CR1 (CD35) and CR2 (CD21), belong to the “regulators of complement activation” (RCA) family.35,36 CR1 (which primarily binds C3b and C4b) is widely expressed by neutrophils, monocytes/macrophages, B lymphocytes, some T lymphocytes, glomerular podocytes, and erythrocytes,37–39 where it has the important function of binding and clearing immune complexes.23,24 CR1 also serves as a cofactor for Factor I, thereby playing a role in regulation of complement activation. Factor I cleaves C3b and C4b to iC3b and iC4b.40,41 CR2 (which is expressed mainly on B lymphocytes, activated T lymphocytes, and follicular dendritic cells) binds primarily iC3b, C3dg, and C3d.42–46
CR2, along with its cognate complement ligands, is a critical link between the innate and adaptive immune systems.26,47 For example, when CR2 (as part of the CD19/CD21/CD81 B-cell coreceptor complex) and B-cell receptors are co-ligated on the surface of B lymphocytes (which occurs via the binding of C3d-decorated immune complexes) B-cell activation, proliferation, and antibody production are enhanced.47–49 Furthermore, CR2 (which is expressed on follicular dendritic cells) can bind to antigens and immune complexes that when they have been opsonized by C3-derived fragments can be retained in the germinal centers of secondary lymphoid follicles.43,44 These retained antigen fragments provide essential signals for generation of memory B-cells, as well as for survival and affinity maturation of B-cells.50
CR3 and CR4 belong to the β2 integrin family and are composed of two subunits: a common β chain (CD18) and a specific α chain (CD11b for CR3 and CD11c for CR4). Phagocytic cells (e.g., neutrophils, monocytes, and macrophages), antigen-presenting cells (e.g., dendritic cells), and follicular dendritic cells express these receptors.51,52 CR3 and CR4 mediate phagocytosis of C3-opsonized pathogens and adhesion of mononuclear phagocytes to endothelial cells.53–55
COMPLEMENT AND SLE
The role of complement in the etiopathogenesis of SLE is complex, intriguing, and paradoxical, and has been intensely studied over the past several decades. On the one hand, a hereditary deficiency of a component of the classical pathway (C1, C2, or C4) has been associated with the development of SLE.5–7,59–61 On the other hand, activation of the complement system [caused by tissue deposition of immune complexes (formed from autoantigens and autoantibodies)] results in tissue inflammation and damage.56–58 Studies performed over the past several years may reconcile seemingly discordant roles for complement in SLE. These studies have demonstrated that although the complement system plays a role in maintaining immune tolerance to prevent the development of SLE59,61,62 it also participates in tissue-destructive inflammatory processes once SLE is established in a patient.56,57
Immune Complex Abnormalities, Complement Activation, and SLE
Immune complex abnormalities (e.g., decreased solubility and impaired disposal of immune complexes) and consequent complement activation are responsible for many of the clinical manifestations and pathology observed in patients with SLE. Decreased serum levels of C3 and C4 (due to genetic or acquired factors, or both) may prevent the formation of small soluble immune complexes63,64 by not permitting sufficient binding of C3 and C4 fragments to the antigen-antibody lattice. Furthermore, impaired binding, processing, and transporting of immune complexes to phagocytes of the reticuloendothelial system may be the result of reduced levels of CR1 on erythrocytes, which have been demonstrated in patients with SLE (see further discussion in material following). As a consequence, insoluble aggregates (which are formed from abnormally large quantities of immune complexes in the circulation) may eventually be deposited in various tissues.
Deposited immune complexes, although they do not seem to cause tissue damage directly, do so indirectly by providing ample binding sites for complement components. The ensuing activation of the complement system causes the release of various mediators and promotes cellular infiltration and interaction, both of which culminate in tissue damage. This pathogenic sequence underlies the molecular basis for changes to the vascular endothelium and glomerular basement membrane, both of which are highly susceptible to this mode of inflammatory damage and lead to vasculitis and glomerulonephritis (two hallmark manifestations of SLE).
Complement Deficiency and SLE
The intriguing clinical association between complement deficiency and SLE is currently explained by three non mutually exclusive hypotheses. In the first hypothesis, impaired clearance of immune complexes due to deficiency of early complement components may trigger/augment the development of SLE. Interestingly, of the two isotopes of human C4, C4A has predominantly been implicated in binding and solubilizing immune complexes.65–67 Consequently, reports that the prevalence of C4A deficiency is higher in SLE patients than in the general population are not unexpected.68–70 Several studies showed impaired initial splenic processing of immune complexes in SLE patients,71–74 supporting the concept that impaired clearance of immune complexes may contribute to the development of SLE in the context of complement deficiency.
The second hypothetical mechanism suggests that complement deficiency, through its capacity to determine activation thresholds of B and T lymphocytes, may alter the normal mechanism of negative selection of self-reactive lymphocytes.26,62,75–77 Co-ligation of CR2 and BCR augments B-cell activation by decreasing the threshold to antigenic stimulation.47 Therefore, self-antigens not opsonized by C4b or C3b are unlikely to trigger sufficient activation of self-reactive B-cells, and as a result these cells may escape negative selection. The escaped cells may breach self-tolerance to autoantigens once they become activated by encountering relevant autoantigens in the periphery.
A third hypothetical mechanism also proposes to explain the link between complement deficiency and development of SLE.78,79 This hypothesis was first generated by the discovery that C1q can bind directly to apoptotic keratinocytes, and has been supported by subsequent observations. These studies demonstrated that endothelial cells and peripheral blood mononuclear cells undergoing apoptosis also bind C1q80 (Fig. 20.3), which subsequently triggers activation and deposition of C4 and C3 on these apoptotic cells.80,81 Thus, opsonized apoptotic cells and blebs can be effectively taken up by phagocytic cells via a complement receptor-mediated mechanism.81,82
During apoptosis, normally hidden intracellular constituents are often biochemically modified and redistributed/segregated into surface blebs of dying cells, resulting in the presence of “altered-self” constituents.83–85 When apoptotic cells are not promptly cleared due to complement deficiency, these constituents may persist, be recognized by the immune system, breach immune tolerance, and trigger autoimmune responses.86 Taken together, these studies suggest that complement plays a pivotal role in maintaining immune tolerance by facilitating the clearance of autoantigen-containing apoptotic bodies.7,59,61,87
Using a mouse model, Botto and colleagues were the first to report that mice deficient in C1q were defective in clearing apoptotic cells, as indicated by the accumulation of apoptotic bodies in the glomeruli of their kidneys. This led to autoimmune responses to nuclear autoantigens and glomerulonephritis.88 Subsequently, Taylor and colleagues reported similar but less severe defects in the clearance of apoptotic cells and spontaneous autoantibody production in C4-deficient mice.89 Results from these animal studies suggest that clearance of apoptotic cells is impaired or delayed in the absence of C1q, and less so in the absence of C4. Likewise, the persistence of apoptotic cells may lead to development of autoimmunity and tissue damage in humans.
Reduced phagocytic activity of neutrophils, monocytes, and macrophages of SLE patients has previously been observed.90–92 Specifically, Hermann and colleagues reported a reduced capacity of SLE-derived macrophages to phagocytose apoptotic cells.93 Moreover, monocyte-derived macrophages prepared from SLE patients have shown impaired clearance of iC3b-opsonized apoptotic cells in vitro.94 Baumann and colleagues reported an abnormal accumulation of apoptotic cells accompanied by a significantly decreased number of tangible body macrophages (cells responsible for removing apoptotic nuclei) in the germinal centers of lymph nodes in a small subset of SLE patients, demonstrating in vivo evidence for impaired clearance of apoptotic cells in human SLE.95 Collectively, data from both animal and human studies not only substantiate the observed hierarchic role for complement components of the classical pathway and the risks of developing SLE but provide a strong mechanistic basis linking complement deficiency and SLE.
CR1 Deficiency in SLE
Reduced levels of CR1 expressed on erythrocytes of patients with SLE have been reported by several investigators.96–108 Studies performed in the 1980s led some investigators to conclude that erythrocyte CR1 deficiency is inherited,96,98,99,102 whereas later studies using SLE patients of various ethnic backgrounds suggested that erythrocyte CR1 deficiency is acquired.97,100,101,103–110 Some studies showed that reduced E-CR1 levels correlated with disease activity.100,104,107,108 Differing experimental methods and ethnic populations may account for these conflicting results.
Wilson and colleagues examined E-CR1 levels in SLE patients and their relatives to elucidate the molecular basis of the observed differential erythrocyte CR1 expression in SLE patients versus healthy individuals.98 They found that a significant number of patients’ relatives had reduced E-CR1 levels. Three patterns of E-CR1 levels (high, intermediate, and low) could be identified in both SLE patients and family members used as controls. These studies indicated that E-CR1 levels were genetically regulated in a bialleleic co-dominant manner. Later, the same investigators demonstrated the presence of two HindIII-digested CR1 gene fragments, 7.4 kb and 6.9 kb in length, by using DNA probes for the CR1 gene and restriction fragment length polymorphism (RFLP) studies.111 They showed that the 6.9-kb fragment (L allele) was associated with low E-CR1 levels, a heterozygous 7.4-kb/6.9-kb (H/L) pattern correlated with intermediate E-CR1 levels, and the 7.4-kb fragment (H allele) correlated with high E-CR1 levels.
These results are consistent with the originally proposed bialleleic co-dominant fashion of inherited E-CR1 expression.98 However, it should be pointed out that E-CR1 levels were considerably lower in SLE patients than in healthy individuals with the matched HindIII RFLP pattern, suggesting that additional genetic or nongenetic factors may influence E-CR1 expression in SLE patients.102 Although these researchers initially postulated that increased prevalence of the HindIII L/L genotype may be associated with SLE, subsequent studies have not supported their theory. Reports from several studies do not indicate a significant difference in gene frequencies of HindIII H and L alleles between SLE patients and normal controls.101,103,106–108 This suggests that the L/L genotype does not increase susceptibility to SLE.
Two other studies support the notion that low E-CR1 levels in SLE patients are acquired. A 1987 study demonstrated that normal erythrocytes transfused into SLE patients lost significant amounts of CR1 within a few days after transfusion.112 A 1999 study found that CR1 levels on the youngest form of erythrocytes (reticulocytes) of SLE patients were equivalent to those on reticulocytes of normal individuals and that SLE patients lost significantly greater amounts of E-CR1 in the peripheral circulation than did normal controls.113
Abnormal Expression of CR2 and CR3 in SLE
It has been reported that expression of CR2 on lymphocytes of patients with SLE was approximately 50 to 60% lower than those of healthy controls.114–116 Whether or not decreased CR2 expression correlates with increased activity of SLE (and if so, whether it is a cause or a result) is unclear. Nonetheless, animal studies using MRL/lpr mice have shown that decreases in CR1/CR2 expression preceded the development of clinically apparent disease,117 suggesting that decreased CR1/CR2 expression may be involved in the initiation or progression of autoimmune disease.
The role of CR2 in the development and pathogenesis of SLE has been investigated using gene-knockout mice. In mice, CR2 and CR1 are encoded by the same gene through differential splicing of the RNA transcripts.118 Therefore, CR2-deficient (Cr2−/−) mice generated by gene-targeting techniques are also deficient in CR1. Initial studies have shown that Cr2−/−mice on the 129/B6 hybrid background did not develop an autoimmune phenotype.119,120 However, when back-crossed with B6/lpr mice (a mouse model of SLE) Cr2−/−/lpr mice on the 129/B6 hybrid background exhibited more aggressive disease featuring marked splenomegaly and lymhadenopathy, increased ANA and anti-dsDNA titers, and increased immune complex deposition in renal glomeruli at earlier time points than did B6/lpr mice.75
In comparison, Cr2−/−/lpr mice on the B6 background developed autoantibodies but not other manifestations.121 Recent studies of SLE susceptibility genes using the NZM2410 mouse model further support an important role of CR2 in the development of autoimmune phenotypes.122 Boackle and colleagues recently identified Cr2 as a candidate gene within the NZM2410 Sle1c locus.123,124 These investigators showed that B6 mice congenic for the Sle1c locus expressed dysfunctional CR1 and CR2 proteins, which are encoded by a defective Cr2 gene with a single nucleotide mutation. Although these mice developed autoantibodies to chromatin, they did not develop glomerulonephritis. Collectively, these results indicate that the Cr2 gene alone is insufficient to induce expression of a full spectrum of autoimmune phenotype/disease in lupus-prone mice, which apparently requires the contribution of additional SLE susceptibility genes.
Reduced expression of CR3 on lymphocytes of patients with active SLE has also previously been reported.125,126 Immune vasculitis and panniculitis, arthritis, serositis, nephritis, and associated prominent cutaneous changes are some of the clinical manifestations seen in these patients. Although leukocyte adhesion deficiency, which is a congenital deficiency of CR3, is associated with increased susceptibility to pyogenic bacterial infections127,128 the pathophysiologic significance of decreased CR3 expression in SLE remains to be elucidated. Seemingly to the contrary, one study has even shown increased levels of CR3 on neutrophils of patients with SLE.129 Paradoxically, the highest neutrophil CR3 levels were detected in patients with the most severe disease, and in some of these patients the increased CR3 levels on neutrophils returned to normal when disease flares subsided. Researchers thus postulated that neutrophils, in response to complement activation, increase surface expression of CR3 in patients with active SLE. These activated neutrophils may contribute to endothelial injury in SLE.129
MEASURING COMPLEMENT
< div class='tao-gold-member'>
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


