Chapter 47 Rehabilitation of Patients with Neuropathies
Anatomy and Physiology
The peripheral nervous system is made up of 12 cranial nerves and 31 spinal nerves, which innervate specific sensory distributions (dermatomes) (Figure 47-1) and muscle groups (myotomes). In the cervical and lumbosacral regions, the spinal nerves intermingle to create plexuses, which then form individual peripheral nerves. In disease, the distribution of motor and sensory abnormalities characterizes particular spinal nerve, plexus, or peripheral nerve disorders.
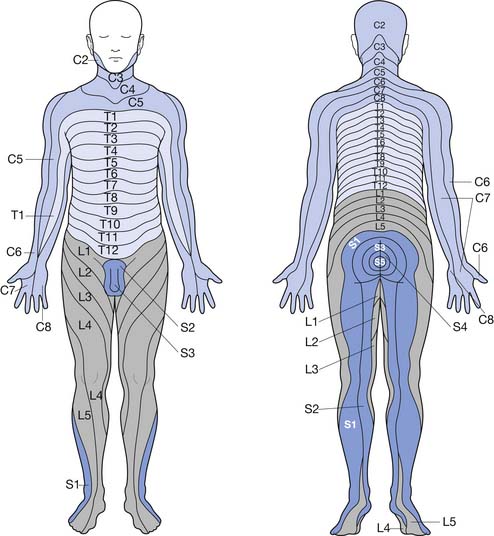
FIGURE 47-1 Dermatomal innervation of spinal nerves.
(Modified from Hockberger RS, Kaji AH: Spinal injuries. In Marx JA, editor: Rosen’s emergency medicine, ed 6, Philadelphia, 2006, Mosby.51)
The basic neural structure is the neuron and its associated axon, or nerve fiber. The axon is enclosed within a Schwann cell. A Schwann cell can encircle multiple axons, in which case they are referred to as unmyelinated. An axon that is invested solely by a Schwann cell wrapping around it several times is known as a myelinated fiber. The myelinated axon is invested by a series of Schwann cells that are separated by small sections of uncovered axon known as nodes of Ranvier (Figure 47-2). Nerve depolarization can “jump” from node to node, a process known as salutatory conduction. Impulse propagation in this manner is far more rapid (50 to 60 m/s) than that in unmyelinated axons (1 to 2 m/s).
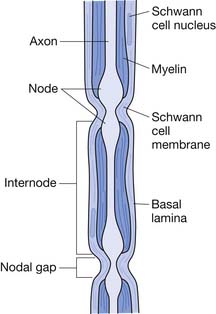
FIGURE 47-2 Myelinated axon invested by Schwann cells separated by nodes of Ranvier.
(Modified from Jobe MT, Martinez SF: Peripheral nerve injury. In Canale ST, Beaty JH, editors: Campbell’s operative orthopaedics, ed 11, Philadelphia, 2009, Mosby.64)
Each nerve fiber and its associated Schwann cells are surrounded by an endoneurium. Several of these axons are grouped together in fascicles that are enclosed in a perineurium (although individual axons can cross from one fascicle to another along the course of a nerve). The fascicles are bundled together within the epineurium, which sheaths the entire peripheral nerve (Figure 47-3).
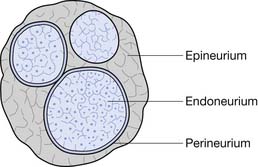
Classification of Neuropathies
Generalized peripheral neuropathies can be characterized by their location along three major axes based on whether the process affects (1) the axon (axonal) or the myelin (demyelinating) or has features of both; (2) the motor or sensory fibers; or (3) the peripheral nerves in a symmetric (diffuse) fashion, typically in a distal to proximal gradient, or in an asymmetric or multifocal pattern (Box 47-1). Neuropathies can also preferentially affect specific nerve types, such as large fiber or small fibers, the latter often including autonomic involvement. For example, alcoholic neuropathy tends to be predominantly axonal, sensorimotor, and diffuse, whereas classic Guillain-Barré syndrome (GBS) is demyelinating, with greater motor than sensory involvement and a multifocal pattern.
BOX 47-1 Classification of Peripheral Neuropathies and Common Etiologies
Localized nerve injuries, or mononeuropathies, are classified by the degree of axonal and supporting structure involvement. The main classification schemes are the Seddon system123 and the Sunderland system.135 Neurapraxia in the Seddon system refers to focal injury to the myelin causing conduction block without axonal injury. Axonotmesis in the Seddon system refers to axonal damage with resultant Wallerian degeneration, but with the supporting endoneurium and perineurium still intact. Neurotmesis is the most severe insult and involves injury to the axon, myelin, and supporting structures of the nerve (typically the nerve is no longer in anatomic continuity). In the Sunderland system, first-degree injury and second-degree injury correspond to neurapraxia and axonotmesis, respectively. The Sunderland system subdivides neurotmesis into third-degree injury (which involves the axon and endoneurium), fourth-degree injury (which affects the perineurium as well), and fifth-degree injury (which reflects damage to all supporting structures of the nerve) (Table 47-1).
Table 47-1 Classification of Nerve Injury: Seddon and Sunderland Systems123,135
| Seddon | Sunderland | Description |
|---|---|---|
| Neurapraxia | First degree | Focal conduction block without axonal damage |
| Axonotmesis | Second degree | Axon damage with wallerian degeneration, supporting structures intact |
| Neurotmesis | Third degree | Damage to axon and endoneurium |
| Fourth degree | Damage to perineurium and endoneurium | |
| Fifth degree | Damage to axon and all supporting structures |
Evaluation of Generalized Neuropathies
History
Neuropathies can be associated with many medical comorbidities, including diabetes, renal failure, and HIV. Possible toxic exposure should be investigated, as well as search for any medications that might be associated with neuropathy (Box 47-2). A careful family history might reveal known hereditary neuropathies or similar symptoms and findings, such as gait problems or pes cavus, suggesting a previously undiagnosed familial condition.
Physical Examination
A general inspection can reveal clues to the presence of a neuropathic condition. Atrophy might be noted, especially involving the intrinsic muscles of the hands and feet. Foot deformities, such as pes cavus, can be seen in those with hereditary neuropathies. Other foot deformities such as hammer toes and even deformity and collapse of the midfoot architecture can be seen in advanced neuropathic disease. Autonomic dysfunction can result in decreased sweating and dry, cold feet. Large fiber neuropathy can result in increased blood flow and “hot foot.”150 Skin lesions or ulceration might be seen in an insensate foot. Fasciculations are occasionally observed.
Muscle stretch reflexes are lost in a distal to proximal gradient in the setting of a diffuse neuropathy, whereas deviation from this pattern suggests a multifocal process. The Achilles reflex is the most commonly affected in a diffuse neuropathy and can be obtained either with direct percussion over the tendon or by plantar strike technique, which might be more reliable in older persons.100 Facilitation might be needed, such as gentle plantar flexion. Distinguishing the “normal” distal decrement in neuromuscular function associated with aging from that resulting from disease can be difficult. The physical findings that are predictive of electrodiagnostic confirmation of peripheral neuropathy in this population is loss of the Achilles reflex, inability to perceive at least 8 of 10, 0.5- to 1.0-cm movements, or being unable to detect a vibrating 128-Hz tuning fork for at least 8 seconds at the great toe.108
Motor deficits can also be found in a distal to proximal gradient, or in a multifocal distribution, and are often characterized by easy fatigability. Detection of weakness might be improved by testing the muscle in question multiple consecutive times.88 Pain, poor cooperation, and poor understanding of examiner instruction can interfere with full muscular effort. In such circumstances, the examination is augmented by observing the patient performing functional tasks. For example, unipedal stance time is a sensitive test of balance impairment. Not only is a markedly decreased unipedal stance time associated with a diffuse peripheral neuropathy in young and middle-aged men,60 but unipedal stance time also strongly correlates with frontal plane sensory and motor function at the ankle among older persons with neuropathy.46,130 Clinicians should observe the patient walking and look carefully for increased variability of lateral foot placement and frank crossover steps. These suggest instability during single-limb stance and increase the risk of foot collisions during ambulation. The distal upper extremity sensorimotor function can similarly be evaluated by assessing the ability to do fine motor tasks, such as buttoning a shirt without visual input.
Electrodiagnostic Studies
Nerve conduction studies and needle electromyography (EMG) are useful in detecting, characterizing, and assessing the severity of peripheral neuropathy. Electrodiagnostic studies build on what is learned after a thorough physical examination. Sensory nerve conduction studies assess the number of axons excited and the speed of conduction of the axons. The amplitude, measured from baseline to peak amplitude, correlates with the number of axons excited,29 and a decrement usually suggests axon loss. In a demyelinating process, dysynchrony of the axons measured can cause temporal dispersion of the sensory response, with decreased amplitude without true axon loss. Reduced sensory amplitude with relatively normal distal latency and conduction velocity suggests axon loss. If the sensory nerve response is completely absent, it is not possible to characterize whether it is an axonal or demyelinating process; consequently an intact sensory response elsewhere should be sought.
Motor nerve conduction studies assess the amplitude of the compound muscle action potential (CMAP), distal latency, and the conduction velocity using proximal and distal stimulation sites. Reduction in CMAP usually reflects axon loss; however, it can also be seen when stimulating across areas of demyelination. Focal demyelination can be found by stimulation proximal and distal to the site, with stimulation at the proximal site resulting in lower amplitude, temporal dispersion, and conduction slowing compared with the distal site. In severe demyelination the action potential can fail to conduct entirely, referred to as conduction block (see Chapters 9 through 11). Acquired demyelinating neuropathies often present with nonuniform demyelination and focal conduction block, whereas hereditary neuropathies usually result in uniform demyelination but without focal block. Late responses (F waves) are useful for assessing more proximal segments and can be particularly sensitive for demyelinating processes, such as early GBS.19
Neuropathies caused by demyelination are defined by the following criteria: (1) conduction velocity slowed to less than 80% of the lower limit of normal (LLN) if the CMAP is more than 80% of LLN and less than 70% if CMAP is less than 70% of LLN; (2) distal latency is prolonged more than 125% of the upper limit of normal (ULN) if CMAP is more than 80% of LLN and more than 150% of ULN if CMAP is less than 80% of LLN; (3) F-wave latency more than 120% of ULN if CMAP amplitude was more than 80% of LLN and more than 150% of ULN if CMAP is less than 80% of LLN, or absence of F-wave response; and (4) partial conduction block with a more than 30% decrease in amplitude between stimulation of proximal and distal sites.2 Amplitudes are generally preserved, although some decrease can be seen as a result of conduction block. Axonal neuropathies will result in amplitude loss without significant prolongation of distal latency or conduction slowing; however, fastest conducting fibers can be preferentially affected, resulting in mild slowing.
Although nerve conduction studies are the most useful component of the electrodiagnostic examination in the evaluation of neuropathies, it is important to be attentive to their limitations. For example, nerve conduction studies predominantly test large fiber nerves and so will likely be unrevealing in pure small fiber neuropathies. It is also important to be aware of conditions that can distort the results, such as temperature of the limb, superimposed focal neuropathies such as carpal tunnel syndrome (CTS), and chronic pressure on foot intrinsic muscles as occurs with diminution of tibial motor amplitude responses in the setting of overpronated feet. Studies should assess at least one motor and one sensory nerve in the upper and lower limb. If abnormalities are found, the contralateral side should be examined to document symmetry. If a multifocal neuropathy is suggested from history and physical examination, a more extensive study is required, assessing the clinically affected nerves. Late responses (F waves) should be obtained, particularly if a demyelinating process is suspected. If a mild distal neuropathy is clinically suspected but sural sensory responses are normal, the plantar sensory responses might reveal abnormalities.1,107
Complications of Neuropathies
Foot Complications
Foot ulceration is a major complication in patients with peripheral neuropathy, particularly in those with diabetes mellitus, who manifest a lifetime risk of 15%.38 The presence of neuropathy is associated with an 8- to 18-fold increased risk of foot ulcer and a 2- to 15-fold greater risk of amputation.101 Loss of protective sensation in peripheral neuropathy can lead to unrecognized foot trauma and skin breakdown. Motor neuropathy causes muscle atrophy and weakness, distorting normal foot architecture, and leading to abnormal pressure distribution and overloaded plantar areas.
Autonomic dysfunction causes decreased sweating, resulting in skin dryness and cracking, further compromising skin integrity. The detrimental impact of autonomic dysfunction is accentuated by arteriovenous shunting, which leads to alteration of perfusion with secondary osteopenia caused by bone resorption and impaired healing. In addition to skin breakdown, neuropathy can also lead to Charcot changes. Charcot neuroarthropathy is characterized by destruction of the joints of the foot, with pathologic fracture and joint dislocation often in the midfoot region, and can result in severe deformity (Figure 47-4).
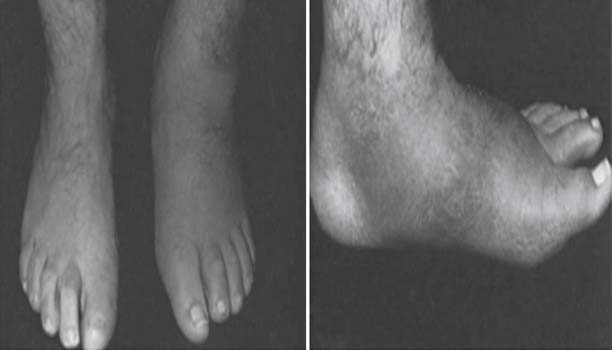
FIGURE 47-4 Charcot neuropathy causing pathologic fracture, dislocation, and deformity of the midfoot.
(From Ishikawa SN: Diabetic foot. In Canale ST, Beaty JH, editors: Campbell’s operative orthopaedics, ed 11, Philadelphia, 2009, Mosby.61)
Patients with neuropathy should be educated on the importance of daily inspection of their feet using a mirror. Properly protective socks and appropriate shoe wear, including extra-depth toe boxes and custom orthoses, should be used to offload high pressure areas. Feet should be kept dry and the skin well moisturized, with caution to avoid extreme temperatures, particularly hot water. Nail care and callus management is important and might necessitate care by a podiatrist or diabetic foot clinic. Prompt management of skin breakdown and aggressive management of infection are important to avoid progression to amputation. Acute Charcot foot can present similarly to cellulitis, as a warm, erythematous, and swollen foot. Radiographs can be normal acutely, before showing the fractures and joint dislocation later in the course of the disease. The bone scan, however, is often positive early. Strict immobilization with a total contact cast is used to manage acute Charcot arthropathy.149 Immobilization is maintained until clinical signs of warmth and tenderness have significantly decreased and the bone scan becomes negative or less active, indicating that bone remodeling is near complete. At this point careful weaning from the orthosis can begin. This is often done in association with physical therapy using a gradual resumption of weight-bearing and strengthening foot and ankle musculature. Prevention and treatment programs can significantly reduce lower extremity breakdowns and amputations.57
Pain
Treatment of neuropathic pain can be challenging, and the side effects of the medications used can be problematic, particularly in elderly patients. A topical agent such as capsaicin cream has the advantage of having no systemic side effects, but the application can be cumbersome (requiring application 3 to 4 times/day), and can cause initial irritation. Transdermal lidocaine patches are more easily applied and can be useful in those with a relatively discrete distribution of pain.25 Transcutaneous electrical stimulation might also be of benefit. Tricyclic antidepressant medications were the first class of medications to show efficacy in the management of neuropathic pain. Amitriptyline’s use is limited by anticholinergic effects, including sedation, dry mouth, urinary retention, and orthostatic hypotension, as well as potentially dangerous arrhythmias.96 Low-dose nortriptyline, dosed at 10 to 50 mg at bedtime, might be better tolerated and has been shown to have equivalent effect on some types of neuropathic pain as amitriptyline153 and with fewer side effects. Gabapentin, at doses up to 3600 mg/day, has been shown to reduce pain.42,124 Gabapentin can cause somnolence and dizziness, particularly on initiation of treatment. It might exacerbate gait and cognitive problems in older patients, but it is generally well tolerated and lacks significant drug interactions. Gabapentin should be initiated at low doses, 100 to 300 mg at night, and slowly titrated up to three times daily dosing. Duloxetine (Cymbalta) and pregabalin (Lyrica) are two newer drugs that have been approved by the Food and Drug Administration for the treatment of neuropathic pain. Duloxetine’s use can be limited by gastrointestinal side effects. Pregabalin, like gabapentin, can cause sedating and cognitive side effects. Tramadol has been shown to have beneficial effects on the allodynia associated with diabetic peripheral neuropathy.128 It binds to μ-opioid receptors and is a weak norepinephrine and serotonin reuptake inhibitor. Tramadol can cause cognitive impairment, and serotonin syndrome might occur with concomitant use of other serotonergic medications. Opioid analgesics have been shown to improve pain and sleep but not function or mood.106 Because opioids can cause cognitive side effects, physiologic dependency, and addiction, they should be used judiciously. Second-line medications for the treatment of neuropathic pain include lamotrigine, carbamazepine, other selective serotonin reuptake inhibitors, and clonidine.
Functional Impairment
Peripheral neuropathy can have significant impact on functional mobility and quality of life.22 Patients with peripheral neuropathy are about 20 times more likely to fall than those without neuropathy.109,113 Neuropathy impairs proprioceptive sensation at the ankle,146 and those with neuropathy are less able to rapidly develop ankle torque to correct for lateral lean.46 Therefore patients with neuropathy are doubly penalized when their center of mass is displaced because it takes a relatively greater displacement before loss of balance is perceived and then there is a delay in the generation of ankle torque to correct. This is particularly exacerbated on uneven surfaces and in dim lighting, where visual cues cannot assist in compensating for proprioceptive loss. Both a perceived and real risk of instability can lead to increasingly limited physical activity and physical deconditioning. Those with neuropathy often become more isolated because of fears of community ambulation on unpredictable terrain and decreased walking speeds, which can be particularly problematic in situations such as crossing the street.
A number of interventions can improve gait and balance. Visual input must be optimized. Vision should be regularly tested, and corrective eyewear should be used as needed. Patients should consider using glasses for walking that correct distance vision only because the use of bifocals has been independently associated with falls.84 Patients should be taught to use proper lighting, particularly when getting up at night. Balance can be improved with proper shoe wear that has a wide base of support and thin soles. A cane can stabilize gait, but to achieve maximal benefit, patients should be able to support up to 25% of their body weight on the cane to prevent a fall.6 The cane should be brought down with each contralateral footstep. Acceptance of the use of a cane can be improved if presented as a substitute for the loss of a special sense, much like eyeglasses, and used only as needed on uneven surfaces and in unfamiliar areas. A disadvantage of canes, however, is that they can be associated with decreased walking speed. Use of ankle orthoses with medial and lateral support has been shown to improve gait and stability parameters.116 They also have the advantage of freedom of both hands and better walking speed than a cane, but the skin in contact with the orthoses must be monitored for breakdown. Although there is no direct evidence that physical training prevents falls, specific exercise programs have shown improvement in clinical measurements of balance in patients with neuropathy.115 Specific exercises aimed at strengthening hip abductors and abdominals can improve hip and trunk stability in the frontal plane. Strengthening of grip, shoulder depression, and elbow extension can improve support when using a cane or other means of upper limb stabilization.
Specific Neuropathies
Diabetic Neuropathies
Chronic Sensorimotor Distal Polyneuropathy
Chronic sensorimotor distal polyneuropathy is the most common type of diabetic neuropathy and affects at least half of those with long-standing diabetes.19 Onset is usually insidious, occurring first in the feet and progressing proximally over time. Symptoms are usually only perceived in the hands when the lower limb symptoms have reached the level of the knees. If upper limb symptoms develop before that time, then an entrapment neuropathy should be suspected. Most patients have involvement of both large and small nerve fibers, although large fiber involvement usually predominates.
Large fiber involvement affects both motor and sensory nerves. Symptoms are often minimal in early stages, with patients describing odd sensations of walking on cotton, vague unsteadiness, or difficulty manipulating small objects such as buttons. Pain is usually less of a complaint but when present is described as a dull, cramping ache. Often these complaints are only elicited with a specific inquiry. Physical examination often reveals abnormalities before symptoms are perceived. Vibratory sensation and position sense loss are some of the earliest findings, along with depressed Achilles reflexes. Muscle wasting, particularly of the intrinsic foot muscles, is found as the neuropathy progresses. Electrodiagnostic studies can demonstrate findings consistent with both axon loss and demyelination. Sensory abnormalities usually occur first, with the sural sensory studies showing the earliest changes on routine studies. If suspicion is high in a patient younger than 60, however, medial and lateral plantar nerves might demonstrate earlier changes (they are more distal than the sural nerve recording).1,107 Motor nerve conduction abnormalities occur later in the course of the disease. EMG needle examination typically shows neuropathic abnormalities in a symmetrical pattern, with a distal to proximal gradient. This often occurs before there is any clinical weakness. With regard to the three descriptive axes mentioned previously, diabetic neuropathy often shows features of both demyelination and axon loss, has greater sensory than motor involvement, and is diffuse.
Involvement of small unmyelinated C fibers can occur early in diabetes and presents with significant pain and hyperalgesia. Later there is loss of thermal sensation and autonomic abnormalities, with loss of sweating, dry feet, and vasomotor changes. This leads to increased risk of foot ulceration and infections. Later in the course of the neuropathy the pain might subside, but this is a sign of progression rather than regression of the disease.150 When small fiber disease occurs in isolation from large fiber involvement, there can be minimal physical examination findings despite significant symptoms. Additionally, electrodiagnostic studies are not as sensitive for small fiber nerves and can be normal.55 Galvanic skin responses might be abnormal but are unreliable.76 Skin biopsy can be definitive, with quantification of small fibers, but is not done routinely.129
Proximal Motor Neuropathy
Diabetic proximal motor neuropathy, also known by the term diabetic amyotrophy, is typically seen in older patients with type 2 diabetes. It classically presents acutely or subacutely with severe pain that is unilateral or bilateral. The pain tends to attenuate over weeks to months, an event often accompanied by marked atrophy of the thigh muscles, particularly the quadriceps, adductors, and iliopsoas, with relative sparing of gluteal and hamstring muscles.80 Sensory abnormalities are seen in the femoral distribution and sometimes in the saphenous distribution. Proximal diabetic neuropathy is almost invariably seen in diabetic patients with preexisting distal neuropathy. A variant of this condition can present with acute and debilitating pain in a younger patient with early, mild diabetes (or previously undiagnosed diabetes), with no evidence of distal neuropathy. Electrodiagnostic studies reveal a pattern of lumbosacral plexopathy sometimes combined with multilevel radioulopathy on EMG needle examination. Femoral nerve conductions reveal predominantly axonal involvement, with reduced CMAP but relatively spared distal latency. Distal nerve conduction studies often show evidence of a concomitant distal neuropathy, even in patients without clinical complaints of distal neuropathic symptoms. The mechanism of injury is thought to be an immune-mediated microvasculitis affecting the nerve roots and plexus. Management of diabetic amyotrophy consists of tight glycemic control and aggressive management of neuropathic pain. Corticosteroids, plasmapheresis, and intravenous immunoglobulin (IVIG) have not been shown to be efficacious in management of this condition. Although extremely debilitating, the prognosis for improvement is good, with recovery typically occurring over a 12- to 24-month period.138 Differential diagnosis includes spinal stenosis and chronic inflammatory demyelinating polyneuropathy (CIDP). Spinal stenosis is common in older adults, and although compression is most common at the L5 level, when occurring at higher levels it can mimic symptoms of diabetic amyotrophy. CIDP is also common in this age group, and it is important to diagnose because it is amenable to steroids, IVIG, plasmapheresis, and other immune-modulating agents.
Focal Mononeuropathies
Mononeuropathies typically occur in older diabetic individuals and are usually acute in onset and associated with pain. Recovery is spontaneous and occurs over 6 to 8 weeks. Cranial nerves III, VI, and VII and median, ulnar, and peroneal nerves are most commonly affected,150 and the neuropathies are caused by microvascular infarction. Patients with diabetes can also be more predisposed to entrapment neuropathies at common sites,65 such as the median, ulnar, and peroneal nerves. Carpal tunnel syndrome is 3 times more common in individuals with diabetes than in the general population.69
Autonomic Neuropathy
Autonomic dysfunction of the gastrointestinal system can lead to esophageal dysmotility, gastroparesis, constipation or diarrhea, and bowel incontinence.12 Gastroparesis can cause a sensation of bloating, nausea, heartburn, early satiety, and erratic glycemic control. These symptoms are typically treated with metoclopramide.87
Diabetic autonomic neuropathy can lead to erectile dysfunction and is usually irreversible.33 Treatment consists of assessing for secondary causes of erectile dysfunction, counseling, medications, suction erectile devices, and penile implants. Neurogenic bladder leads to overflow incontinence, recurrent urinary tract infection, and pyelonephritis. If retention is suspected, it can be diagnosed with postvoid residuals and might need more formal urodynamic studies to determine the severity. In mild cases the patient can be instructed to void every 2 to 3 hours while awake. In more severe cases intermittent catheterization can be necessary.
Guillain-Barré Syndrome
GBS was first described almost a century ago by French neurologists Guillain, Barré, and Strohl. With the eradication of polio, it is now the most common cause of acute neuromuscular paralysis in the Western world, with an annual incidence of 1 to 2 per 100,000.105 GBS is a progressive, symmetrical weakness of the limbs, with hyporeflexia or areflexia, with or without sensory abnormalities. Maximal weakness occurs within 4 weeks, although typically peaks by 2 weeks. GBS is now recognized to be a group of disorders, with various subtypes. The most common is the acute demyelinating polyradiculoneuropathy (AIDP) affecting both motor and sensory nerves. This type accounts for 95% of all cases of GBS in Europe and North America.48 Purely axonal forms are rare (<5%). Axonal forms can occur as an acute motor axonal neuropathy (AMAN) or acute motor and sensory axonal neuropathy (AMSAN). The axonal forms of GBS are more common in Asia and South America, making up 30% of the cases in those populations.102 The Miller Fisher variant is a triad of ataxia, areflexia, and ophthalmoplegia, although bulbar abnormalities, ptosis, facial weakness, and papillary dysfunction can be seen (Table 47-2).37
| Clinical Features | Antibodies | |
|---|---|---|
| AIDP | Anti-GM1 | |
| Axonal (AMAN, ASMAN) | Anti-GM1, GD1a, Ga1Nac-GD1a | |
| Miller Fisher | Anti-GQ1b |
AIDP, Acute demyelinating polyradiculoneuropathy; AMAN, acute motor axonal neuropathy; AMSAN, acute motor and sensory axonal neuropathy.
Filippi Rl, Charalampaki R, Reisch R., et al: Recurrent cubital tunnel syndrome: etiology and treatment, Minim Invasive Neurosurg 44: 197-201, 2001.
Clinical Features
GBS presents with progressive onset of limb weakness both proximally and distally that is typically symmetrical. It reaches its maximal effect in 2 to 4 weeks. Reflexes are typically lost early in the disease, although reflexes can be retained or even brisk with axonal forms.74 Sensory loss is variable. The cranial nerves can be affected, with facial palsy and bulbar weakness. Muscles of respiration are frequently affected with decline of vital capacity, and ventilatory support is required in 25% of cases. The autonomic system can be impaired, causing tachycardia, hypertension, and cardiac arrhythmias. Pain can be a significant complaint and might even precede onset of clinical weakness. In the acute stages the pain is often described as deep and aching, affecting the back, buttocks, and posterior thighs. This might be due to nociceptive pain from inflammation. Later neuropathic pain can be described with degeneration and regeneration of sensory nerves.148 Fatigue is a common complaint, even after good neurologic recovery from GBS. Amantadine and other pharmacologic agents have not been shown to help, but bicycle exercise training has been shown to reduce fatigue and improve functional outcome and quality of life.40
Pathophysiology
Two thirds of patients with GBS have an antecedent infection with 3 weeks of onset of weakness. The most commonly identified infectious agent is Campylobacter jejuni. Other causative agents are cytomegalovirus (CMV), Epstein-Barr virus, Mycoplasma pneumoniae, and Haemophilus influenzae.49 Studies of the swine flu vaccine during the 1976 epidemic demonstrated an increased risk of one excess case of GBS for every 100,000 people vaccinated.122 Little strong evidence is known to link GBS with other vaccines, including newer influenza vaccines.47 The strong association with antecedent infection suggests that at least in some cases there is an immune response to these infectious agents that triggers damage to peripheral nerves. The preceding infectious agent is also associated with the subtype of GBS that develops. Patients with C. jejuni infection are more likely to develop axonal forms of GBS, and those with CMV infections tend to have more cranial nerve, respiratory, and severe sensory involvement. Epstein-Barr virus infection usually confers milder disease.105 The infectious agents share localized regions capable of eliciting an immune response (epitopes) with antigens in peripheral nerve tissue, triggering antibody-mediated damage to the peripheral nerves. Various antiganglioside antibodies have been identified in the subtypes of GBS. Nerves are affected at the root level initially, followed by distal segments, and the intervening areas later in the course of the disease.8
Diagnosis
Required clinical features for the diagnosis of GBS are progressive weakness in both arms and legs, and areflexia. Supportive features include progression of weakness over days to 4 weeks, symmetry of symptoms, mild sensory symptoms, cranial nerve involvement, autonomic involvement, pain (which can be prominent), high protein concentration in the cerebrospinal fluid (CSF), and typical electrodiagnostic findings. A number of features raise doubts about the diagnosis of GBS (Box 47-3).
Electrodiagnostic testing is the most useful confirmatory test for GBS and can differentiate between the more common demyelinating form (AIDP) and axonal forms, AMAN, and AMSAN. Early in the course of AIDP, the nerve conduction studies can be normal. Because the nerve roots are typically affected first, prolongation of the F-wave latencies might be the earliest abnormality seen as they measure conduction in the proximal nerve segments.3 Nerve conduction studies demonstrate a demyelinating pattern with motor involvement occurring earlier and more extensively than sensory abnormalities. Sensory changes tend to be patchier in distribution, and median and ulnar sensory responses tend to be affected to a greater extent than the sural response. Temporal dispersion and partial conduction block, findings consistent with an acquired demyelinating disease, can be seen between proximal and distal stimulation sites. Needle examination typically shows decreased firing of motor units on maximal contraction. An axonal variant of GBS is suggested if there is significant reduction in CMAPs and early and significant findings of abnormal insertional activity on needle examination. CMAPs that fall below 20% of LLN predict poor functional outcome.93 Electrodiagnostic findings usually lag behind clinical severity.
Management of Guillain-Barré Syndrome
Management of GBS consists of supportive care, disease-modifying treatment, and rehabilitation. Most patients require admission to the hospital for close monitoring. Mortality for GBS is 10%, with infections, pulmonary embolus, and cardiac arrhythmia the most common causes of death. Deep venous thrombosis prophylaxis should be initiated, particularly in nonambulatory patients. Commonly used agents include subcutaneous heparin or low-molecular-weight heparin and sequential compression devices. Vital capacity should be sequentially monitored every 2 to 4 hours initially, and mechanical ventilation should be initiated when vital capacity falls below 20 mL/kg.79 Autonomic involvement can result in labile blood pressure and cardiac arrhythmias and can require monitoring in the intensive care unit. Serious bradyarrhythmias can develop, requiring atropine or transcutaneous pacemaker.156 In some patients pain can be difficult to treat, requiring opioids, gabapentin, and other agents for this neuropathic pain. Prevention of pressure ulcers with proper positioning and turning, as well as range of motion and splinting to prevent contractures, should be instituted. Swallowing function should be monitored, particularly in those with signs of facial or bulbar involvement.
Immunotherapy has been proven to be of benefit in patients with GBS. Plasma exchange administered five times over the course of 2 weeks is beneficial when started within 4 weeks of onset of weakness but is most effective within the first 2 weeks.45 IVIG has been shown to be as effective as plasma exchange147 and is usually the preferred treatment because of greater convenience and availability. It is administered at 0.4 g/kg daily for 5 consecutive days. Combining plasma exchange and IVIG has not been shown to be superior to monotherapy.104 Five to 10 percent of patients develop neurologic deterioration after initial improvement after treatment. It is common practice to administer a second course of IVIG in these patients. Oral or intravenous steroids have not been shown to be effective in altering the course or functional outcome of GBS.59
Prognosis of GBS is typically good, especially given the often marked initial degree of weakness. Persistent disability, however, occurs in 20% of patients who will be nonambulatory or require an assistive device to walk at 6 months.58 Poor outcome is associated with advanced age, male gender, axonal involvement, antecedent diarrhea, and CMV infection.105 Relapse is rare, at a rate of 3% to 5%.49
Chronic Inflammatory Demyelinating Polyneuropathy
The electrodiagnostic features of CIDP were first described in 197532; however, since then several variants have been recognized. CIDP is an immune-mediated disorder of the peripheral nervous system. The classic form of CIDP is symmetric neuropathy that affects motor function predominantly, both proximally and distally. Variants include a multifocal, asymmetric form and both a motor and symmetric form known as Lewis-Sumner syndrome. Variants also exist that are purely sensory forms, as well as forms associated with IgG and IgA paraproteins, central nervous system demyelination, and various systemic disorders (Box 47-4). A number of conditions also are important to differentiate from CIDP variants, especially because they respond differently to treatment. These include multifocal motor neuropathy and IgM-related neuropathies such as POEMS (polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes).
BOX 47-4 CIDP Variants
CIDP, Chronic inflammatory demyelinating polyneuropathy; Ig, immunoglobulin.
Clinical Features
CIDP progression occurs over at least 2 months, a feature that differentiates it from GBS. Motor symptoms generally predominate, and both proximal and distal muscles are affected. Because CIDP is a primarily demyelinating disorder, muscle atrophy is not a significant feature. Muscle stretch reflexes are reduced or absent. Cranial involvement can occur in 10% to 20% of cases, and a small number of patients have painful dysesthesias. Sensory involvement typically affects large fiber nerves, consequently preferentially affecting vibration and proprioception. Sensory symptoms generally progress from distal to proximal, although hand involvement is often perceived as early as that in the feet. The classic course of CIDP is one of recurrence and remittance, a pattern that tends to occur in younger patients. Older patient are more likely to have a progressive form of CIDP.121
Diagnosis
Electrodiagnostic studies are essential for confirmation of CIDP. Nerve conduction studies demonstrate primary demyelination in multiple segments. Findings consistent with demyelination include conduction block, conduction slowing that is greater than can be explained by axon loss, prolonged F-wave and H-reflex latencies, and temporal dispersion of CMAP between proximal and distal stimulation points. Sural nerve biopsy can show demyelination or findings pointing to other disorders that can mimic CIDP, such as vasculitis or amyloidosis. Because CIDP is multifocal and motor predominant, however, sural biopsy can be normal and therefore is not routinely done. Lumbar puncture will show aluminocytologic dissociation in 90% of patients.32 Magnetic resonance imaging (MRI) with gadolinium of the brachial and lumbar plexi and cauda equina can reveal enhancing or enlarged nerves.
The European Federation of Neurological Societies/Peripheral Nerve Society proposed diagnostic criteria for CIDP based on review of available evidence and expert concensus.67 Diagnostic probability is based on presence of typical or atypical clinical features (Box 47-5), electrodiagnostic findings of demyelinating disease (Box 47-6), and supportive evidence. Supportive evidence includes MRI abnormalities, CSF findings, nerve biopsy, and response to immunomodulating treatment. These recommendations permit the description of CIDP as typical or atypical, with or without concomitant disease, and definite, probable, or possible.
BOX 47-5 Clinical Criteria of Chronic Inflammatory Demyelinating Polyneuropathy (CIDP)
From Johnson R. K., Spinner M, Shrewsbury MM, Median nerve entrapment syndrome in the proximal forearm, J Hand Surg Am 4: 48 51, 1979.






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


