Chapter 10 Regulatory Cells in SLE
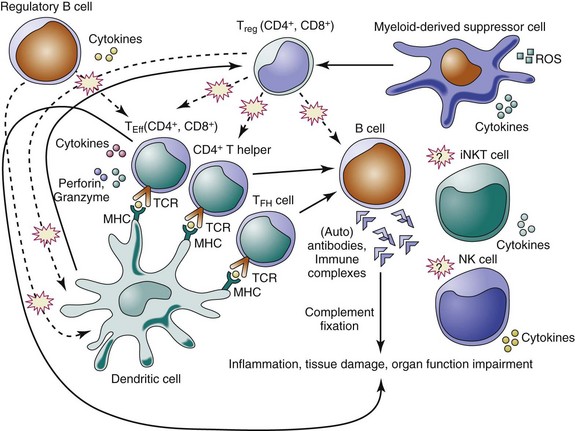
The suppressive activity of regulatory cells appears crucial in preventing autoimmunity, in reducing inflammatory responses caused by pathogens and environmental insults, and in maintaining immune homeostasis. Here we focus on this specific aspect of immune tolerance by discussing the individual subsets of immunoregulatory cells in relation to SLE (Table 10-1; Figure 10-1).
TABLE 10-1 Schematic Summary of the Phenotypic Markers of the Main Subsets of Immunoregulatory Cells in SLE
| CELL TYPE | MOUSE | HUMAN |
|---|---|---|
| Regulatory T cells: | ||
| CD4+ Tregs | CD4+CD25+Foxp3+ | CD4+CD25highFoxP3+CD127− |
| CD4+CD25highFoxP3+ICOS+/− | ||
| Additional markers: CTLA-4, GITR, CD45RBlow, CD62Lhigh, neuropilin-1, CD103, CD5, CD27, CD38, CD39, CD73, CD122, OX-40 (CD134), TNFR2, LAG-3, CCR4, CCR7, CCR8 | ||
| CD8+ Tregs | CD8+CD28− | CD8+CD28− |
| CD8+CD25+Foxp3+ | CD8+CD25+FoxP3+ | |
| CD8+CD122+ | CD8+CD122+ | |
| CD8+CD103+Foxp3+ | CD8+CXCR3+ | |
| CD8+CD27+CD45RA+ | ||
| Additional markers: CTLA-4, GITR, CD44, Ly49 | ||
| Regulatory B cells | CD1dhighCD5+B220+ | CD19+CD24highCD38high |
| CD1dhighCD21highCD23+IgMhigh | ||
| Myeloid-derived suppressor cells | CD11b+GR-1low | |
| Dendritic cells | CD11c+CD103+ | |
| Natural killer cells | NKG2D+, CD56bright | |
| Invariant natural killer T cells | Vα14Jα18/Vβ8.2 | Vα24JαQ/Vβ11 |
GITR, glucocorticoid-induced tumor necrosis factor receptor; LAG-3, lymphocyte activation gene 3; TNFR, tumor necrosis factor receptor.
Regulatory T Cells
CD4+ Regulatory T Cells
CD4+ regulatory T cells (Tregs) are the most-studied subset of immunoregulatory/suppressor cells. These cells help control immune self-reactivity, allograft rejection, and allergy, and they inhibit effector cell functions in infections and tumors. The deficiency or reduction of Tregs in normal mice leads to the development of autoimmune responses because these cells actively suppress the activation and expansion of autoreactive immune cells, and a restoration of the number of Tregs associates with the reversal of autoimmune phenotypes in several experimental animal models.1,2
CD4+ Tregs are generally classified into two main categories, the thymus-derived natural Tregs (nTregs), and the Tregs that can be induced peripherally from CD25− precursors in vivo3—called adaptive or induced Tregs (iTregs) or—in vitro with interleukin-2 (IL-2) and transforming growth factor (TGF)-β4 or IL-10 (the IL-10–producing type 1 Tregs are generally called Tr1 cells).5 Both Treg types suppress CD4+ effector cell activation, proliferation, and cytokine production as well as CD8+ effector cell activation, proliferation, and cytotoxic activity in vitro, and B lymphocytes.6 At present, there are no reliable phenotypic or functional markers that make it possible to reliably distinguish between natural and induced Tregs.
Natural Tregs represent 5% to 10% of peripheral CD4+ T cells in mice and are characterized by the constitutive expression of CD25 (IL-2 receptor α-chain). In humans, they make up 1% to 2% of the peripheral blood CD4+ T cells, particularly the ones with the highest CD25 expression (CD25high or CD25bright).7 However, CD25 is not a unique marker for Tregs because it is also present on activated T cells—and is thus also expressed by effector T cells.8 To discriminate Tregs from conventional (activated) CD4+ T cells, particularly in humans, it may be useful to include additional markers such as a low expression of CD127 (the α chain of the IL-7 receptor)9 and a modulated CD45RB expression, together with the expression of CD25high and the intracellular expression of FOXP3.10 FOXP3 (forkhead box P3) is an X chromosome–encoded member of the forkhead/winged-helix family of transcription regulators whose discovery led to a significant advancement in the understanding of the biology of Tregs.11 Mice and humans harboring a loss-of-function mutation in the FOXP3 gene are affected by fatal lymphoproliferative immune-mediated disease, the IPEX (immune dysregulation, polyendocrinopathy, enteropathy, X-linked) syndrome in humans12 and the scurfy phenotype in mice.13,14 FOXP3 is required for the development, maintenance, and suppressor function of Tregs,11,15 and the loss of FOXP3 in Tregs—or its reduced expression—leads to the acquisition of effector T-cell properties that include the production of non–Treg-specific cytokines.16,17 However, the expression of FOXP3 per se may not be sufficient for a regulatory cell function, because human activated T cells can also express FOXP3 even without possessing a suppressive capacity.18 Yet, FOXP3 remains at present the best marker for the identification of Tregs.
Another marker that discriminates two subsets of thymus-derived FOXP3+ Tregs is the co-stimulatory molecule ICOS,19 which distinguishes ICOS+ Tregs with high IL-10–producing capacity from ICOS− Tregs that produce TGF-β.19 Additional markers that also describe Tregs are the cytotoxic T lymphocyte–associated antigen 4 (CTLA-4), the glucocorticoid-induced TNF receptor (GITR), CD45RBlow and CD62Lhigh expression, neuropilin-1, CD103 (integrin aEβ7), CD5, CD27, CD38, CD39, CD73, CD122, OX-40 (CD134), tumor necrosis factor receptor 2 (TNFR2), lymphocyte activation gene 3 (LAG-3), C-C chemokine receptor type 4 (CCR4), CCR7, and CCR8.20
In regard to Treg development, nTregs originate in the thymus through high-avidity major histocompatibility complex (MHC) class II–dependent/T-cell receptor (TCR) interactions,21–23 with the induction of FOXP3 upon TCR engagement in thymus,24 whereas peripherally, FOXP3 expression appears influenced by factors such as intracellular signaling, cell proliferation, and the synergy with TGF-β and IL-2.25–26 In addition to the TCR, CD28 co-stimulation also seems to play an important role in the differentiation of Tregs, and a marked decrease in the frequency of Tregs is observed in CD28-deficient and CD80/CD86-deficient mice.27,28 Of interest, the CD28/B7 signaling pathway is essential for the development of nTregs but it may not be needed for the development of iTregs (although it promotes their expansion).27 Additionally, the development and function of both nTregs and iTregs appear negatively regulated by OX40, a member of the TNF–TNF receptor superfamily.29,30
IL-2 and TGF-β play an essential role in the differentiation and development of iTregs, and the combination of IL-2 and TGF-β can induce CD25−FOXP3− precursors to express FOXP3 and acquire a suppressive phenotype.3,4 Interestingly, iTregs generated in vivo and Tr1 cells may not express FOXP3, whereas iTregs induced ex vivo by IL-2 and TGF-β typically express FOXP3 and share many phenotypic and functional characteristics with nTregs.25,31,32 In this context, it has been reported that the foxp3 gene locus and its enhancer in nTregs could be structurally distinct from those in iTregs. DNA methylation can affect Tregs’ stability and their suppressive capacity in vitro, and although demethylation of CpG motifs within the foxp3 locus is observed in nTregs, only partial demethylation of CpG motifs is observed in iTregs.33 Furthermore, a specific site within a unique and evolutionarily conserved CpG-rich island of foxp3 upstream enhancer has been found unmethylated in nTregs but not in iTregs.34 It is not known why methylation status in the TSDR (Tregs-specific demethylated region) of iTregs generated in vitro is different from that found in those generated in vivo, but it seems that iTregs induced in vivo have both stable FOXP3 expression and demethylated TSDR.35
The mechanisms of action of human and mouse Tregs have been studied mostly in vitro. Targets of the activity of Tregs include CD4+CD25− T cells, CD8+ T cells, B cells, monocytes, and dendritic cells (DCs).2,20,36,37 Tregs (particularly nTregs) operate through cell-to-cell contact mechanisms that involve the release of cytotoxic molecules, including perforin and granzymes A and B.38 Gene expression arrays showed that granzyme B was upregulated in mouse Tregs,39 and human Tregs expressed granzyme A and lysed activated CD4+ and CD8+ T cells in a perforin-dependent manner.40,41 Tregs could also kill B cells in a granzyme B–dependent and partially perforin-dependent manner,1,42 or could induce apoptosis of effector T cells upon the upregulation of the TRAIL-DR5 (tumor necrosis factor–related apoptosis inducing ligand-death receptor 5) pathway43 or galectin-1.44 Other means by which Tregs can suppress target cells is a metabolic disruption that includes cytokine deprivation–mediated apoptosis, cyclic AMP (cAMP)–mediated inhibition,45 and the expression of the ectoenzymes CD39 and CD73 (which can generate pericellular adenosine, which inhibits activated T cells or inhibits DCs through the activation of the adenosine receptor 2A).46–48
Regarding the cytokines that influence Treg activity, TGF-β seems to play a key role. In vitro studies that used neutralizing antibodies to this cytokine—or that employed Tregs that lacked TGF-β indicated that TGF-β was dispensable for Tregs’ suppressive functions.49,50 However, other studies found a relevant role for cell membrane–tethered TGF-β on Tregs in their suppressive activity, both in vitro and in vivo.51,52
Another newly described inhibitory cytokine, IL-35, which can be expressed by Tregs, might also contribute to their suppressive capacity or could operate on targets.53
Lastly, other mechanisms employed by Tregs can involve direct effects on maturation and function of antigen-presenting cells (APCs) through the expression of CTLA-454,55 and the inhibitory lymphocyte activation gene 3 (LAG-3, or CD223, a CD4 homolog that binds MHC class II molecules with very high affinity).56
CD4+ Tregs and SLE
Lupus-prone mice have a lower frequency of Tregs than non-autoimmune mouse strains.57 Although a deficit of Tregs in murine SLE was found to contribute to the development of the disease in mice,58 adoptive transfer of in vitro–expanded CD4+CD25+CD62Lhigh Tregs slowed the progression of renal disease and decreased mortality in lupus mice.57 However, the effect of adoptive transfer in mice in which proteinuria had already developed was modest.57 The Tregs that could confer protection and increase the survival in mice with established SLE were the iTregs that could prevent the help of T cells to B cells for the production of anti-DNA antibodies, with a resulting inhibition of immune complex glomerulonephritis and proteinuria.59,60
In human SLE, the investigation of the role of Tregs in the disease has sometimes yielded controversial results. Most studies found a reduced frequency of Tregs in SLE, although other studies showed normal or even increased numbers.60 Although a normal suppressive capacity of Tregs has been described in patients with both active and inactive SLE, it seems overall that the number of Tregs is lower in patients with active disease than in patients with inactive SLE and in normal controls, and this lower number would be associated with reduced levels of FOXP3 and a poor suppressive capacity in patients with active disease.61–64 Another consideration is that the finding of a reduced inhibition of autoreactive immune responses in SLE could be associated with a resistance of effector target cells to an otherwise normal activity of Tregs.65
Nothwithstanding these aspects, it is interesting to note that a rise in the numbers of Tregs was observed after rituximab-induced B-cell depletion at the time of B-cell repopulation,66 and that therapy with corticosteroids and/or immunosuppressive agents promoted an increase in the number of functional Tregs.67
CD8+ Tregs
Like CD4+ Tregs, CD8+ Tregs can be classified as either natural or induced. CD8+ nTregs develop in the thymus, whereas iTregs likely arise in the periphery from cells that initially do not express regulatory functions but acquire them after antigenic stimulation. The phenotype of CD8+ nTregs resembles that of CD4+ nTregs, and these cells generally express FOXP3 as well as CD25 in addition to surface CTLA-4 and GITR.68 Other subsets of CD8+ Tregs are CD8+CD28− T cells69; CD8+CD103+FOXP3+GITR+CTLA-4+ T cells induced by allostimulation and facilitated in culture by IL-10, IL-4, and TGF-β70; and CD8+CD122+71 or CD8+ T cells that coexpress CD44 and Ly49 and directly suppress follicular T helper (TFH) cells (and thus autoantibody production) by recognizing Qa-1/peptide complexes on TFH cells and depend on IL-15 for development and function.72
As mechanisms of suppression, CD8+ Tregs employ the secretion of the cytokines IL-10 (used by human CD8+CXCR3+, CD8+CD122+, and CD8+CD27+CD45RA+ Tregs), TGF-β, IFN-γ, and IL-16,73–79 cell-to-cell contact (e.g., in a membrane-bound TGF-β—and in a CTLA-4–mediated, contact-dependent manner ),68 and cytotoxicity (e.g., on activated CD4+ Th cells, which depends on the expression of the MHC class 1b molecule Qa-1 or HLA-E in humans).80–82 CD8+ Tregs could also induce a tolerogenic phenotype in APCs that would in turn favor the induction of CD4+ Tregs.68
CD8+ Tregs and SLE
Murine models of tolerogenic vaccination with peptides have shown that the induction of Tregs (both CD4+ and CD8+ Tregs) protected mice from SLE manifestations.59,77,83–85 Tolerization of (NZB × NZW)F1 (BWF1) mice with the anti-DNA Ig–based peptide pCons expanded CD8+ Tregs capable of suppressing (1) anti-DNA autoantibodies in vivo and in vitro, (2) CD4+ T-cell proliferation, and (3) IFN-γ production.86 These CD8+ iTregs secreted TGF-β and required FOXP3 for their suppressive function.86,87 Their induction was associated with a reduced expression of programmed death 1 (PD-1) molecules on those cells,88 which influenced their immunoregulation capacity.89 In the same BWF1 lupus model, another tolerogenic peptide based on human anti-dsDNA antibodies also induced CD8+CD28− Tregs that suppressed lymphocyte proliferation and autoantibody production, increased TGF-β production, and decreased IFN-γ and IL-10 production as well as lymphocyte apoptosis.85,90,91 Similarly, a histone-derived tolerizing peptide (H471-94) in (SWR × NZB)F1 (SNF1) lupus-prone mice increased survival and decreased anti-dsDNA autoantibodies in lupus mice, expanding CD8+ Tregs that expressed GITR and TGF-β.77 Finally, in a graft-versus-host disease (GVHD) murine model of lupus, both CD4+ and CD8+ Tregs that required IL-2 and TGF-β increased survival.92
In patients with SLE, some studies reported defective and/or reduced numbers of or no difference in CD8+ Tregs in comparison with healthy controls.93–95 One study comparing CD8+ Tregs from SLE patients and healthy controls generated by culture of CD8+ T cells with IL-2 and granulocyte-monocyte colony-stimulating factor (GM-CSF) showed that CD8+ Tregs from patients with active SLE could not suppress effector T cells, whereas CD8+ Tregs from patients whose SLE was in remission had a suppressive capacity similar to that of cells from healthy controls.93 In another study on the effects of autologous hematopoietic stem cell transplantation in patients with refractory lupus, patients who showed clinical improvement after transplantation had an increase in the number of CD4+ and CD8+ Tregs, including the CD8+CD103+ T-cell susbset.96
Regulatory B Cells
Certain autoantibodies can be found in healthy individuals, but in autoimmune settings these antibodies can cause tissue damage through local inflammation that ultimately leads to impaired organ function. B cells are key contributors to the pathogenesis of SLE, not only because they make autoantibodies but also because they can present self-antigens and because they secrete cytokines that can sustain or amplify the autoimmune response. On the other hand, it has become apparent that certain subsets of B cells can also exert immunoregulatory functions and contribute to the inhibition of autoimmune responses. In mice, the regulatory function of B cells is almost exclusively dependent on IL-10.97 The cell surface phenotype of murine regulatory B cells is typically CD1dhighCD5+ or CD1dhighCD21highCD23+IgMhigh, and thus it overlaps with that of CD5+ B-1a cells, CD1dhighCD21highCD23lowIgMhigh marginal zone (MZ) B cells, and CD1dhighCD21highCD23highIgMhigh transitional type 2 (T2)–MZ precursor B cells.98 As such, a regulatory function for B cells seems to be present in MZ B cells, T2-like B cells, and CD5+ B cells.
In comparison with mouse regulatory B cells, less is known about human regulatory B cells: It seems that human CD19+D24highCD38high B cells have regulatory capacity.99
The initial suggestion that B cells could exert immunoregulatory functions came from studies in mice in which the depletion of B cells abolished the inhibition of skin inflammation.100 Subsequent studies showed that B cells could have immunoregulatory functions in humans and in several murine animal models, and some underlying mechanisms of action have been elucidated. The activation of regulatory B cells seems to require three signals: BCR, CD40, and Toll-like receptors (TLRs).101 CD19 has also been found to be important in the development of regulatory B cells.102–104 Genetic deficiency of CD19 resulted in an increased and prolonged inflammation in autoimmune-prone mice, whereas overexpression of CD19 associated with the expansion of regulatory B cells.104 For CD40, signaling through this molecule expressed on B cells was required for regulatory B-cell development, and CD40 appeared to be involved in the regulatory mechanisms used by B cells.105 In this context, B cells also express the ligand for CD40 (CD40L), which makes possible an autonomous B-cell control of IL-10 production (the production of IL-10 in CD40L+ B cells correlates with CD40L expression levels in some autoimmune diseases).104,106
Currently the following two models are proposed for the B cell–mediated immunoregulation of effector CD4+ T cells, Tregs, invariant natural killer T (iNKT) cells, and DCs: (1) a direct regulation due to cell interactions or the secretion of soluble factors and (2) an indirect regulation via effects on intermediate cells. For example, regulatory B cells could suppress APC function by producing IL-10 or C-X-C motif chemokine 13 (CXCL13) or could downregulate CD4+ T-cell responses by engaging their TCRs.107 Regulatory B cells could also activate iNKT cells through an increased CD1d expression.108,109 The regulatory effects could involve CD40 or B7 co-stimulatory molecules for the mechanisms involving cell-to-cell contacts98 or soluble factors (i.e., the B cell–derived IL-10, considering that B cells from IL-10–deficient mice cannot protect from autoimmunity98 and that activated B cells in the presence of neutralizing anti-IL-10R fail to exert regulatory functions110).
B cells that produce IL-10 include peritoneal CD5+ B-1a cells, CD5−CD11c−CD21+ B cells in Peyer patches, and lupus CD21+CD23− MZ cells (in response to CpG stimulation).111–113 A B-2–like phenotype (CD5−CD11b−IgD+) of IL-10–producing regulatory B cells detectable after IL-7 stimulation has also been identified.114 Subsets of B regulatory cells that produce TGF-β have also been described, suggesting that certain B cells could use TGF-β to inhibit Th1 autoimmunity (by inducing apoptosis in Th1 cells) and/or by inhibiting antigen presentation98,115 or inducing CD8+ T-cell anergy.116 Another suppressive mechanisms used by B cells could be the secretion of antibodies, because under certain circumstances, antibodies can contribute to the downregulation of inflammatory responses and participate in immunoregulation by binding the Fcγ receptor FcγRIIB on DCs to suppress APC function.117,118 Incidentally, passive administration of antibodies associated with the reversal of inflammation in B cell–deficient mice119 and beneficial effects in some patients with SLE.120
Regulatory B Cells and SLE
In one study, low-dose CD20 monoclonal antibody (mAb) treatment in BWF1 mice at 12 to 28 weeks of age followed by administrations every 4 weeks delayed SLE, whereas B-cell depletion initiated in 4-week-old mice hastened the onset of disease concomitantly with the depletion of IL-10–producing regulatory B10 cells.121 In another study, CD19-deficient BWF1 mice had delayed development of antinuclear antibodies in comparison with wild-type BWF1 mice, but showed pathologic signs of lupus nephritis much earlier and had reduced survival, indicating both disease-promoting and protective roles for B cells in the pathogenesis of SLE.122 Also in the second study, IL-10–producing regulatory B cells (CD1dhighCD5+B220+) were increased in wild-type BWF1 mice and were deficient in CD19-deficient BWF1 mice, and the transfer of these cells from wild-type animals into the CD19-deficient ones prolonged the latters’ survival.122
In humans, CD19+CD24highCD38high regulatory B cells were found to suppress the differentiation of Th1 cells after CD40 stimulation in the presence of IL-10 but not TGF-β, and their suppressive capacity was reversed by blockade of CD80 and CD86.123 Also, CD19+D24highCD38high from patients with SLE were refractory to further CD40 stimulation, produced less IL-10, and lacked the suppressive capacity of their healthy counterparts.123
Myeloid-Derived Suppressor Cells
Myeloid-derived suppressor cells (MDSCs) are a heterogeneous population of cells that expands during the course of inflammation, infection, and cancer.124 MDSCs include immature granulocytes, monocytes/macrophages, certain DCs, and early myeloid progenitors, and in mice they express CD11b and GR-1.125 The mechanisms of suppression used by these cells include the production of arginase-1 (which depletes the target cells of L-arginine), the formation of nitric oxide and reactive oxygen species (ROS),126 and the induction of CD4+ Tregs.127 Certain macrophages also display similar suppressive capacities associated with the production of IL-10 and the capacity to influence tryptophan catabolism in target cells in addition to modulating levels of ROS and L-arginine. For example, macrophages stimulated with M-CSF (macrophage colony-stimulating factor) were found to express indoleamine 2,3-dioxygenase, which reduced tryptophan availability and inhibited T-cell proliferation.128
Myeloid-Derived Suppressor Cells and SLE
In MRL(lpr-lpr) lupus mice, CD11b+GR-1low cells were found to suppress CD4+ T-cell proliferation, which was restored by the arginase-1 inhibitor nor-NOHA. These MDSCs regulated immunologic responses via signaling by chemokine (C-C motif) ligands CCL2/CCR2.129
In a chronic graft-versus-host disease model of lupus, Csf3r was identified as the causative gene of the lupus-susceptible Sle2c2 interval in NZM2410 lupus mice that was used by MDSCs in the suppression of T cells.130
Dendritic Cells
DCs are found in multiple tissues, including the gut, lung, skin, internal organs, blood, lymphoid tissues, and bone marrow and may display different functions that depend on the tissue microenvironment.131 Schematically, DCs can be classified into conventional DCs and pre-DCs (which need further differentiation into DCs). Conventional DCs are further classified into migratory DCs (which move to draining lymph nodes) and lymphoid tissue–resident DCs, which capture the antigen in lymphoid organs. In addition to these conventional DC subtypes, a DC population that produces large amounts of type I IFNs is represented by the plasmacytoid DCs (pDCs). Migratory and lymphoid tissue–resident DCs can be classified into subtypes. For migratory DCs the classification is based on the tissue of origin, whereas for lymphoid tissue–resident mainly DCs it is based on specific markers.132–134 For example, mouse skin contains two populations of langerin+ DCs: epidermal Langerhans cells (LCs) and dermal DCs (DDCs). The dermis also contains migratory LCs and langerin− DC. The skin draining lymph nodes contain different DC populations that express CD11c: CD8+DEC205+ resident DCs, CD8−DEC205− (both CD4− and CD4+ resident DCs), CD8lowCD205int DCs (migratory DDCs), and CD8lowDEC205high DCs (migratory LCs).135 Under homeostatic conditions, DDCs and LCs continuously migrate to draining lymph nodes,136 and the spontaneous migration of the DCs to lymph nodes in steady-state conditions contributes to the maintenance of immune tolerance to tissue antigens.137,138 Other DCs are found in Peyer patches or in the lamina propria in the gut (where they produce IL-10 and perform local immunoregulatory functions).139 DCs at these locations could be responsible for maintaining tolerance to commensal bacteria and food.140,141
The immunoregulatory function of DCs in the gut has been attributed to a CD103+ DC subpopulation that efficiently mediates the conversion of naïve T cells into iTregs. Studies in vitro have shown that CD103+ DCs isolated from the lamina propria of the small intestine and from mesenteric lymph nodes can induce iTregs’ differentiation in the presence of TGF-β and retinoic acid, the active derivative of vitamin A.142
It is thought that conventional DCs can be tolerogenic if antigen presentation occurs in the absence of inflammation, through mechanisms that could involve apoptosis, anergy, and Treg activation and expansion.143 In this regard, the conversion of naïve CD4+ T cells into iTregs has been attributed to migratory DCs reaching the skin-draining lymph nodes and displaying a semimature phenotype.144 In one study, DC-mediated expansion of Tregs appeared to be contact-dependent and required IL-2 and the expression of B7 co-stimulatory molecules.145 Studies with CD40-deficient mice also showed that DCs helped maintain Treg homeostasis through cell-cell contact, CD40-CD40L interaction, and IL-2 production.146 Of interest, DCs could tolerize not only CD4+ T cells but also CD8+ T cells via the cross-presentation of exogenous antigens and the involvement of inhibitory molecules such as PD-1 and CTLA-4.147
Typically, DCs that have presented their antigens to T cells are eliminated by apoptosis,148 so that in physiologic conditions DCs die by apoptosis 48 hours after activation.149 Significant accumulation of DCs was observed in patients with autoimmune lymphoproliferative syndrome type II (who have a defect in apoptosis)150 and in lpr mutant mice (DC apoptosis may be Fas-dependent or Fas-independent),151 suggesting that defects in DC apoptosis might contribute to autoimmunity.
Dendritic Cells and SLE
Low-dose tolerance achieved with the histone-derived peptide H471-94 prolonged the lifespan of lupus mice and effectively induced CD4+ and CD8+ Tregs that suppressed autoreactive Th and B cells and renal inflammation.77 In investigating these findings, it was found that the peptide H471-94 induced a tolerogenic phenotype in splenic DCs that captured the peptide, facilitated the production of local TGF-β, and allowed DC-mediated induction of Tregs together with the inhibition of Th17 cells that infiltrated the kidney of the lupus mice.152 Tregs could also be induced in SLE by mature human monocyte-derived DCs that expressed indoleamine 2,3-dioxygenase (IDO).153 Other DC-mediated effects on SLE were found to be secondary to immune complexes/Ig that inhibited DC maturation and enhanced tolerogenicity of DCs (through the engagement of FcγRIIb and the induction of prostaglandin E2).154

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


