Raynaud’s Phenomenon and Systemic Sclerosis
Laura B. Hughes
Barri Fessler
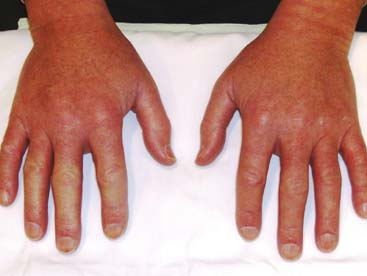 |
A 42-year-old man presents complaining of swollen hands. His fingers turn blue and white, and are associated with pain when exposed to cold temperatures. He notes heartburn and the sensation of food sticking in his esophagus. Examination reveals diffusely edematous hands with skin thickening affecting the fingers. Nail-fold microscopy shows dilated loops with areas of dropout (Fig. 12.1).
Raynaud’s Phenomenon
Introduction
Raynaud’s phenomenon (RP) is an exaggerated vasospastic response to cold temperature or emotional stress. First described by Maurice Raynaud in 1862 it is characterized by intermittent acral bleaching, followed by cyanosis and erythroderma. The typical tricolor sequence is driven by vasoconstriction of digital arteries (white phase), decreased blood flow in capillaries and venules (blue phase), followed by reactive hyperemia (red phase). Population-based surveys estimate the prevalence of RP in women between 6% and 20% and men between 3% and 12.5% (1).
 Figure 12.1 Skin thickening of both hands. Pallor of the 2nd, 3rd, and 4th digits of the right hand. |
Clinical Presentation and Examination
Raynaud’s phenomenon occurs as a primary (not associated with an underlying disease) or secondary syndrome (associated with an underlying disease) (2). The distinction between primary and secondary RP is important as their pathophysiology differs, and the prognosis, severity, and treatment may also differ (3). Primary RP is characterized by the following diagnostic criteria: a definite history of symmetric episodic attacks of acral pallor or cyanosis; absence of peripheral vascular disease; absence of tissue necrosis; normal nail-fold capillary examination; a negative antinuclear antibody (ANA) test; and a normal erythrocyte sedimentation rate (4). Patients with primary RP often are younger and have minimal pain with attacks. In patients with secondary RP, the course is often more
severe and frequently results in ischemic changes and digital ulceration (4). Many connective tissue diseases are associated with secondary RP, most notably systemic sclerosis (SSc), where it is often the initial manifestation. It may also occur in systemic lupus erythematosus (SLE), myositis, Sjögren’s syndrome, rheumatoid arthritis, mixed connective tissue disease, vasculitis, and undifferentiated connective tissue disease. Secondary RP can also occur in noninflammatory conditions including hand–arm vibration syndrome, thoracic outlet syndrome, occlusive vascular diseases (e.g., arteriosclerosis, atheroemboli, thromboangiitis obliterans), hematologic diseases (e.g., paraproteinemia, cryoglobulinemia, cryofibrinogenemia, cold agglutinin disease, polycythemia), and from medications (e.g., amphetamines, beta-blockers, cocaine, nicotine, antineoplastic agents) (5).
severe and frequently results in ischemic changes and digital ulceration (4). Many connective tissue diseases are associated with secondary RP, most notably systemic sclerosis (SSc), where it is often the initial manifestation. It may also occur in systemic lupus erythematosus (SLE), myositis, Sjögren’s syndrome, rheumatoid arthritis, mixed connective tissue disease, vasculitis, and undifferentiated connective tissue disease. Secondary RP can also occur in noninflammatory conditions including hand–arm vibration syndrome, thoracic outlet syndrome, occlusive vascular diseases (e.g., arteriosclerosis, atheroemboli, thromboangiitis obliterans), hematologic diseases (e.g., paraproteinemia, cryoglobulinemia, cryofibrinogenemia, cold agglutinin disease, polycythemia), and from medications (e.g., amphetamines, beta-blockers, cocaine, nicotine, antineoplastic agents) (5).
Clinical Points
Raynaud’s phenomenon may occur as a primary (i.e., not associated with an underlying disease) or secondary syndrome (i.e., associated with an underlying disease); making this distinction is important because prognosis differs.
The diagnosis of RP is made clinically; no tests are needed.
Scleroderma may be a systemic or localized disease. The most common forms of SSc are the diffuse and limited subtypes that are distinguished on the basis of the extent of skin thickening.
Limited scleroderma typically develops over several decades; diffuse scleroderma evolves rapidly over 1 to 2 years.
Clinical clues to suggest secondary RP include later age of onset, asymmetric finger involvement, intense pain, tissue necrosis, signs or symptoms of another disease (e.g., alopecia, rash, sicca symptoms, oral ulcers, photosensitivity, skin thickening, arthralgias, dyspnea, gastroesophageal reflux disease (GERD), muscle weakness), and abnormal nail-fold capillaroscopy (5). Nail-fold capillaries can be examined through a drop of oil using an ophthalmoscope set at 40 diopters. The presence of enlarged or tortuous capillary loops suggests an underlying connective tissue disease, whereas these findings in association with capillary dropout are more suggestive of SSc.
If a thorough history and physical examination, including nail-fold capillaroscopy, reveals little evidence for an underlying disease, a clinical diagnosis of primary RP can be made. If there is clinical suspicion of a secondary cause, or an abnormal nail-fold capillary pattern is observed, serologic testing should be performed, including an ANA and erythrocyte sedimentation rate. An abnormal nail-fold capillary pattern in a patient with RP has been found to be the best predictor of an eventual disease transition to secondary RP. Elevated titers of ANA antibodies including anticentromere, antinucleolar, or anti-ScL70 antibodies, in a patient with RP, suggest the presence of—or eventual development of—an underlying connective tissue disease.
Studies
Raynaud’s phenomenon is a clinical diagnosis. If secondary causes are suspected, an evaluation to asses for atherosclerotic disease is indicated as well as for an underlying connective tissue disorder, including serologies for SLE, Sjögren’s syndrome, and an autoimmune myositis.
Treatment
Treatment choices for RP depend on the severity of the condition and the presence of an underlying disease. The goals of therapy are to improve quality of life and prevent tissue injury. In patients with primary RP, a conservative, nonpharmacologic approach is most important, although medications may be necessary. General education regarding the disease itself as well as the use of nonpharmacologic lifestyle modifications is recommended. Avoiding unnecessary cold exposure or sudden temperature changes such as moving from a hot environment to an air-conditioned room is essential. Patients should understand that the entire body and not just the digits should be kept warm. Strategies such as wearing thermal underwear, hats, scarves, and insulated footwear help keep the body warm. The digits should be protected from cold with gloves and/or hand warmers. Patients should avoid medications that promote vasoconstriction, such as decongestants, amphetamines, beta-blockers, and caffeine. Similarly, smoking cessation is also recommended because nicotine is vasoconstrictive. Physical maneuvers that promote vasodilation in the digits can also be taught to lessen the severity of an attack, including rotating
the arms in a windmill pattern and placing the hands in warm water or in a warm body fold (such as the axilla). If these measures fail to improve the quantity and/or severity of attacks, there are a number of pharmacologic therapies that can be initiated. Calcium channel blockers are the most widely used class of drugs for the treatment of RP. Among the different classes of calcium channel blockers, the dihydropyridine group has been the most effective, with doses of nifedipine ranging from 30 to 180 mg daily or amlodipine from 5 to 20 mg daily. The long-acting or slow-release preparations are generally preferred as they are better tolerated and achieve a more sustained response. If a patient has a suboptimal response to maximum-dose calcium channel blockers, the addition of a direct vasodilator—such as topical nitroglycerin—can be used. Indirect vasodilators have also been evaluated, including angiotensin converting enzyme (ACE) inhibitors (e.g., enalapril, captopril), angiotensin II receptor antagonists (e.g., losartan), and selective serotonin reuptake inhibitors (e.g., fluoxetine). More recently, phosphodiesterase type 5 inhibitors (e.g., sildenafil, tadalafil, vardenafil) have been used for patients with severe RP with digital ischemia. Bosentan, an endothelin 1 receptor antagonist, has demonstrated success in treating digital ulcers in patients with scleroderma and secondary RP. Prazosin, a sympatholytic agent, and pentoxyphilline, a phosphodiesterase inhibitor, have also been reported to improve RP symptoms. Digital or thoracic sympathectomy or intravenous prostaglandin infusions (e.g., iloprost, epoprostenol) can be utilized in patients with RP who are refractory to oral medical therapy, typically in the acute setting where there is critical digital ischemia. Low-dose aspirin has also been recommended in patients with digital ischemia.
the arms in a windmill pattern and placing the hands in warm water or in a warm body fold (such as the axilla). If these measures fail to improve the quantity and/or severity of attacks, there are a number of pharmacologic therapies that can be initiated. Calcium channel blockers are the most widely used class of drugs for the treatment of RP. Among the different classes of calcium channel blockers, the dihydropyridine group has been the most effective, with doses of nifedipine ranging from 30 to 180 mg daily or amlodipine from 5 to 20 mg daily. The long-acting or slow-release preparations are generally preferred as they are better tolerated and achieve a more sustained response. If a patient has a suboptimal response to maximum-dose calcium channel blockers, the addition of a direct vasodilator—such as topical nitroglycerin—can be used. Indirect vasodilators have also been evaluated, including angiotensin converting enzyme (ACE) inhibitors (e.g., enalapril, captopril), angiotensin II receptor antagonists (e.g., losartan), and selective serotonin reuptake inhibitors (e.g., fluoxetine). More recently, phosphodiesterase type 5 inhibitors (e.g., sildenafil, tadalafil, vardenafil) have been used for patients with severe RP with digital ischemia. Bosentan, an endothelin 1 receptor antagonist, has demonstrated success in treating digital ulcers in patients with scleroderma and secondary RP. Prazosin, a sympatholytic agent, and pentoxyphilline, a phosphodiesterase inhibitor, have also been reported to improve RP symptoms. Digital or thoracic sympathectomy or intravenous prostaglandin infusions (e.g., iloprost, epoprostenol) can be utilized in patients with RP who are refractory to oral medical therapy, typically in the acute setting where there is critical digital ischemia. Low-dose aspirin has also been recommended in patients with digital ischemia.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


