Chapter 26 Pathogenesis and Treatment of Atherosclerosis in Lupus
Introduction
Premature atherosclerosis is a major comorbid condition in systemic lupus erythematosus (SLE). Although typical features of SLE, such as nephritis and vasculitis, have been the traditional focus of treatment, the identification of comorbid conditions such as atherosclerosis has become more important as the treatments for SLE improve and patients live longer. In a landmark study, the higher risk of cardiovascular disease in SLE was first recognized in 1976 by Urowitz, who described a bimodal pattern of mortality in a Toronto SLE cohort.1 Of the 11 deaths in the cohort, 6 deaths occurred within 1 year of diagnosis and were attributed to active SLE disease. Five patients died at a mean of 8.6 years, all of whom had had a recent myocardial infarction (MI), with 4 of the5 deaths attributed to fatal MI.1 This bimodal pattern of mortality due to cardiovascular disease has been confirmed in subsequent studies2 and appears to have remained constant despite improvements in overall lupus mortality. For example, the mortality rate from atherosclerosis in patients with SLE has been between 6% and 16% in various later series.3 Data from a large international cohort suggests that although standardized all-cause mortality rates for SLE decreased from 4.9 in 1970 through1979 to 2.0 in 1990 through 2001, the standardized all-cause mortality rates for cardiovascular disease in lupus did not decrease over the same period.4
The overall prevalence of clinical coronary heart disease is also elevated in patients with SLE and has ranged from 6% to 10% in various cohorts.5–7 This risk is higher than that in the general population; for example, in a Swedish lupus population, the risk of coronary artery disease (CAD) in patients with SLE was ninefold higher than in the age-matched general population.8 The age of onset of cardiovascular disease in SLE also appears to be younger that in the general population; Manzi found that women with SLE in the 35- to 44-year age group were more than 50 times more likely to have an MI than women of similar age in the Framingham Offspring Study.5 Cardiovascular events may also result in greater morbidity and mortality in patients with SLE; such patients have higher risk of in-hospital mortality and prolonged length of hospitalizations than both diabetic patients and patients without SLE and diabetes.9
Subclinical Measures of Atherosclerosis
Our ultimate goal as treating physicians is to detect increased risk of cardiovascular disease in our patients prior to the onset of cardiovascular events, so that treatment strategies can be initiated to prevent morbidity and mortality. The detection of subclinical atherosclerosis using surrogate measurements can predict cardiovascular morbidity and mortality in the general population.10 Using a variety of surrogate measurements, several groups have also found that the incidence of subclinical atherosclerosis is increased in patients with SLE. In a cross-sectional study using carotid ultrasound as a surrogate measure, Roman found that carotid plaque was present in 37% of patients with SLE, compared with 15% of controls.11 In a short-term longitudinal follow-up study of the patients with SLE in this cohort, atherosclerosis developed or progressed at an average rate of 10% per year. Further studies using carotid plaque as a surrogate measure have reflected similar prevalences12–14 and rates of progression15 of subclinical atherosclerosis in SLE. Electron-beam computed tomography (EBCT) has also been used as a screening instrument; in one study, coronary calcification was present in 31% of patients with SLE but only 9% of controls.11 In a study using dual-isotope single-photon emission computed tomographic (SPECT) myocardial perfusion imaging, 38% of patients with SLE had perfusion defects indicating subclinical atherosclerosis.16 When endothelial dysfunction, another marker of subclinical atherosclerosis, was evaluated by ultrasound in another study, 55% of patients with SLE had impaired flow-mediated dilation, compared with 26.3% of control subjects.17
Evidence also exists that in addition to abnormalities of the macrovasculature in SLE, there is abnormal coronary microvascular function as well. In one study, abnormal coronary flow reserve (CFR) (measured by means of positron emission tomography [PET]) was seen even in patients with SLE whose coronary arteries were normal.18 Further, evidence has now revealed a 44% prevalence of abnormal stress myocardial perfusion as shown by adenosine stress cardiac magnetic resonance imaging (MRI) in the absence of obstructive CAD in patients with SLE and angina chest pain; quantitative myocardial perfusion reserve index (MPRI) was observed to be lower in patients with SLE than in controls, and the presence of SLE was a significant predictor of myocardial perfusion reserve index.19 It should be noted, however, that although these measures of subclinical atherosclerosis are significantly linked to coronary events in the general population,10 only abnormal myocardial perfusion has been shown to predict future cardiovascular events in SLE.16
Traditional and SLE-Specific Risk Factors for Atherosclerosis in SLE
Traditional Risk Factors
The traditional Framingham cardiac risk factors—hypertension,3,6,7 hypercholesterolemia,1,5,7 diabetes mellitus,1,7 older age,3,5,7 tobacco use, and postmenopausal status3,5—have all been associated with atherosclerotic disease in patients with SLE (Table 26-1). Assessment of cardiovascular risk factors in the Hopkins Lupus Cohort demonstrated that 53% of patients with SLE had at least three traditional risk factors.7 Although the frequency of some traditional risk factors, like diabetes and hyperlipidemia, may be increased as secondary effects of glucocorticoid therapy,20 there is also evidence that traditional risk factors may be directly influenced by SLE disease activity. For example, high levels of very-low-density lipoprotein (VLDL) and triglycerides and low levels of high-density lipoprotein (HDL) have been described as the “lupus pattern,” and are more strikingly noted in patients with active disease.21
TABLE 26-1 Traditional and Nontraditional Cardiac Risk Factors in Patients with SLE
| RISK FACTOR | STUDIES DEMONSTRATING SIGNIFICANT ASSOCIATION WITH OVERT CLINICAL OR SUBCLINICAL ATHEROSCLEROSIS | STUDIES DEMONSTRATING NO ASSOCIATION |
|---|---|---|
| Traditional Risk Factors | ||
| Age | 5-7,11,12,14,28,37,116,185,186 | |
| Body mass index | 37 | 11,14 |
| Association with increased intima-media thickness (IMT) in children: 36 | ||
| Diabetes mellitus | 6,186 | |
| Dyslipidemia | 13 37 | 11,14 |
| Homocysteine | 12 28 | |
| Hypertension | 6,7,17,35,37,187,188 | |
| Menopausal status | 5 | |
| Tobacco Use | 35 | 11,14 |
| Nontraditional (SLE-Specific) Risk Factors | ||
| Corticosteroid therapy | Inverse association: 11 | |
| High and low doses with increased IMT: 36 13,29,34,37,44 | ||
| Moderate (0.15-0.4 mg/kg/day) doses with decreased IMT in children: 36 | ||
| Renal disease | 28 27,33,34,36 | 37 |
| SLE disease activity | Higher disease activity: 11-13,26,28 | 12,16,27 |
| Lower disease activity: 13 | ||
| SLE duration | 27, 12,13,26 | 189 |
| SLE damage | 12,13,29 | 189 |
Although traditional cardiac risk factors clearly play a role in the pathogenesis of atherosclerosis in SLE, they do not fully explain the increased risk. For example, after data were controlled for gender, blood pressure, diabetes, cholesterol, smoking, and left ventricular hypertrophy in a Canadian cohort, Esdaile found the relative risk attributed to SLE for MI was 10.1 and 7.9 for stroke.22 In a separate cohort, Chung found that 99% of patients with SLE were identified as having low risk using the Framingham risk calculator, with a 10-year risk estimate of less than 1%; however, EBCT demonstrated coronary calcium in 19% of patients with SLE in the cohort.23 Similarly, in an SLE cohort from Toronto, the mean Framingham 10-year risk of a cardiac event did not differ in 250 patients with SLE and 250 controls.24 This study did, however, reveal a higher prevalence of nontraditional cardiac risk factors in patients with SLE, including premature menopause, sedentary lifestyle, and increased waist-to-hip ratio.24 Thus, although patients with SLE are subject to the same traditional risk factors as the general population,22,25,26 these factors do not adequately account for the significantly higher level of cardiovascular disease.
SLE-Specific Risk Factors
Disease Activity, Duration, and Damage
The association between SLE disease activity and atherosclerosis has been poorly understood to date. Romero-Diaz reported that higher mean disease activity scores were significantly associated with increased coronary calcium scores27; however, Manzi found an inverse relationship between SLE activity and carotid plaque,13 and several other groups found no association between disease activity and progression of atherosclerosis.12,28 The association between atherosclerosis and disease duration and damage in SLE has been more consistent; several cross-sectional cohort studies have observed significant associations between longer disease duration and either carotid plaque13 or coronary calcium scores.27,29 In a UK study, Haque found that subjects with clinical coronary heart disease were more likely to have higher SLICC (Systemic Lupus International Collaborative Clinics) damage index scores than matched patients with SLE without CHD.30 Similarly, Roman found through multivariate analysis that longer disease duration and higher SLICC damage index scores were independent predictors of carotid plaque in both a cross-sectional study11 and a longitudinal study.12
Renal Disease
Renal disease also appears to be a risk factor for atherosclerosis in patients with SLE; in one large study, both pediatric and adult patients with end-stage renal disease (ESRD) and SLE had significantly higher mortality due to cardiovascular disease than age-matched patients with ESRD but no SLE.31 In another study of renal transplant recipients with SLE, 82% had evidence of coronary calcium on EBCT.32 Active renal disease, including proteinuria33,34 and elevated serum creatinine,35,36 has been associated with early atherosclerosis in patients with SLE. A history of previous nephritis has also been associated with subclinical atherosclerosis in some29,35,37 but not all studies.11,38 Although the exact mechanisms of the greater cardiovascular disease in patients with SLE nephritis has not been indentified, hypertension39 and dyslipidemia40 may contribute to the increased risk, because both are frequently seen in patients with proteinuria. Patients with proteinuria also have an increased risk of thrombosis.41,42
Glucocorticoid Therapy
Glucocorticoid use may impact traditional cardiac risk factors such as hypertension, obesity, and diabetes.43 Additionally, prednisone doses higher than10 mg/day have been shown to independently predict hypercholesterolemia in SLE.7 Conflicting data exist, however, regarding the overall risk of glucocorticoid therapy: Both longer duration of corticosteroid treatment13,44 and a higher accumulated corticosteroid dose13,30,35,38,45 have been associated with a higher incidence of atherosclerosis in various cohorts of patients with SLE. In the APPLE (Atherosclerosis Prevention in Pediatric Lupus Erythematosus) study of pediatric lupus patients, however, the highest and lowest cumulative doses of corticosteroids were associated with increased carotid artery intima media thickness (IMT), and moderate doses were associated with decreased carotid artery IMT.37 Roman also found that former or current use of prednisone and average dose of prednisone were significantly less in patients with carotid plaque,11 implying that there may be a threshold dose at which the antiinflammatory effects of glucocorticoids may be atheroprotective, whereas higher doses may be atherogenic.
Antiphospholipid Antibodies
Given the strong relationship of antiphospholipid antibodies (aPLs) with arterial and venous clotting complications in patients with SLE, there has been considerable interest in aPLs’ possible involvement in accelerated atherosclerosis. However, the role of aPLs in the development of atherosclerosis in patients with lupus remains controversial. Interestingly, there have been numerous reports of aPLs linked with atherosclerosis in non-lupus populations. Antiphospholipid antibodies have been associated with an increased risk of MI in men in several studies46,47; however, the presence of anticardiolipin antibodies (aCLs) in the Physician’s Health Study was not linked to an increased risk of ischemic stroke.48 In a population-based, case-control study in a population of Dutch women younger than 50 years, lupus anticoagulant positivity, although seen in only 3% of women with MIs and 17% with ischemic stroke on the basis of a single determination, was associated with an increased risk of (MI (odds ratio [OR] = 5.3) and ischemic stroke (OR = 43.1) In this study, anti–beta 2 glycoprotein I (β2-GPI) was associated with an increased risk of stroke but not MI, whereas the presence of aCLs did not increase the risk of either stroke or MI.49
There has also been variability in the association between aPLs and subclinical atherosclerosis. Two small case-control series using carotid ultrasound in antiphospholipid syndrome (APS) did not find increased plaque prevalence.50,51 However, four case-control studies have found that IMT is greater in patients with APS than in healthy controls,52–54 and one case-control study also found impaired flow-mediated dilation in patients with APS.55
Likewise, the associations with aPLs in patients with SLE have been variable. Several studies have demonstrated an association between the presence of aPLs and atherosclerosis in SLE. In the LUpus in MInorities, NAture versus nurture (LUMINA) study, aPLs were an independent risk factor for a cardiovascular or cerebrovascular event after a subject’s entry into the cohort.56 In a univariate analysis, the lupus anticoagulant was the only antiphospholipid associated with MI in the Hopkins Lupus Cohort.57 However, neither lupus anticoagulant (LAC) nor aCL was associated with subclinical atherosclerosis as assessed by carotid ultrasound.58 In another study, coronary calcification scores were associated with aPL positivity in a univariate analysis; however, the association was no longer significant when data were adjusted for age and sex.14 Three studies using carotid ultrasound to detect atherosclerosis did not find any association of plaque with aPL.11,13,59 In a later study of an inception cohort of 1249 patients with SLE who had 22 atherosclerotic events, there was no association of aPLs with the events.60
Animal studies evaluating the role of aPLs in atherosclerosis likewise remain contradictory. George and colleagues immunized LDL receptor–deficient mice with β2-GPI and found that fatty streak formation was accelerated.61 The group were also able to accelerate atherosclerosis by passively transferring β2-GPI–reactive lymphocytes. In a follow-up study, the researchers fed β2-GPI to LDL receptor–deficient mice to induce oral tolerance. They were able to show that the mice that received the β2-GPI had less atherosclerosis and that the oral tolerance was successful as assessed by a inhibition of lymph node proliferation to β2-GPI in these mice.62
Other investigators, however, have suggested the aPLs may play a protective role in the pathogenesis of atherosclerosis. Immunization of rabbits,63 LDL receptor–deficient mice,64 and apolipoprotein E (apo E)–deficient mice65 with LDL and/or oxidized LDL (ox-LDL) inhibited the progression of atherosclerotic lesions. Furthermore, passive administration of a monoclonal immunoglobulin (Ig) G cardiolipin–reactive antibody to LDL receptor–deficient mice reduced plaque formation.66 Further studies are needed to explore the contributions of antiphospholipid antibodies to atherosclerosis.
Novel Biomarkers/“Non-traditional” Cardiac Risk Factors
Inflammation and the Pathogenesis of Atherosclerosis
For many years, atherosclerosis was regarded as a passive accumulation of lipids in the vessel wall. It has been realized, however, that inflammation plays a role not only in the development of the atherosclerotic lesion but also in the acute rupture of plaques that occurs during acute myocardial ischemic events.67 As in the pathogenesis of SLE itself, the interplay of multiple inflammatory mediators, including leukocytes, cytokines, chemokines, adhesion molecules, complement, and antibodies, results in the formation of atherosclerotic plaques.67
Monocyte and T-Cell Recruitment to the Arterial Wall
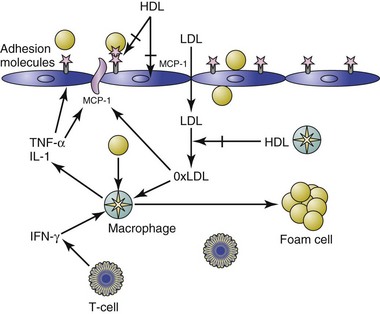
Changes in the vascular endothelium can accelerate the formation of the atherosclerotic plaque. For example, in response to hemodynamic stress, as in cases of hypertension,68 or when exposed to inflammatory mediators such as ox-LDL or cytokines such as interleukin-1 (IL-1) and tumor necrosis factor (TNF), the vascular endothelium undergoes a series of inflammatory changes, resulting in endothelial cell activation (ECA).67 When ECA occurs, there is an up-regulation of leukocyte adhesion molecules such as vascular cell adhesion molecule 1 (VCAM-1), intercellular adhesion molecule 1 (ICAM-1), and E-selectin.68 Chemoattractant cytokines such as monocyte chemoattractant protein 1 (MCP-1), IL-6, and IL-8 are also expressed,68 thus inducing a cascade of proinflammatory, proatherogenic changes in the endothelium that results in migration of monocytes into the subendothelial space. T cells are also recruited to the subendothelial by similar mechanisms, although at lower numbers. These T cells are generally T-helper 1 (Th1) CD4+ cells that secrete proinflammatory and proatherogenic interferon gamma (IFN-γ).67
Low-Density Lipoproteins and the Development of Foam Cells
Next, LDLs are transported in a concentration-dependent manner into artery walls, where they become trapped and seeded with reactive oxygen species (ROS) produced by nearby artery wall cells.69 These LDL phospholipids become oxidized (ox-LDL), and in turn stimulate endothelial cells to release cytokines such as MCP-1, macrophage colony-stimulating factor (M-CSF), and GRO, resulting in further monocyte binding, chemotaxis, and differentiation into macrophages.67 HDL cholesterol is capable of inhibiting the transmigration of leukocytes in response to ox-LDL.70 The ox-LDLs are phagocytized by infiltrating monocytes/macrophages, which then become the foam cells around which atherosclerotic lesions are built.69
Monocytes and T cells infiltrate the margin of the plaque formed by foam cells. Muscle cells from the media of the artery are stimulated to grow.67 These muscle cells encroach on the lumen of the vessel and ultimately lead to fibrosis, which can render the plaques brittle. The occlusion that results in MI can occur when one of these plaques ruptures or when platelets aggregate in the narrowed area of the artery.67
HDL Clears Ox-LDL from the Endothelium
There are many mechanisms designed to clear ox-LDL from the subendothelial space, such as macrophage engulfment using scavenger receptors and enhanced reverse cholesterol transport mediated by HDL.71 Both HDL and its major apolipoprotein constituent, apo A-I, have also been shown to prevent and reverse LDL oxidation.71 In addition to apo A-I, HDL contains several enzymes that can prevent or destroy the formation of the oxidized phospholipids in ox-LDL that induce the inflammatory response; these include paraoxonase, platelet-activating factor acetylhydrolase (PAF-AH), and lecithin : cholesterol acyltransferase (LCAT) (Figure 26-1).71 HDLs are also capable of inhibiting the expression of cell surface adhesion molecules.71
Thus, it is not solely the amount of HDL present that determines atherosclerotic risk, because HDL function is equally significant.71 However, during the acute-phase response, such as in the postoperative period or during influenza infection, HDLs can be converted from their usual antiinflammatory state to proinflammatory (piHDL). In piHDL, levels of antiinflammatory components of HDL such as apo A-I and HDL-associated paraoxonase activity are reduced.72 Additionally, acute-phase HDL is greatly enriched in acute-phase reactants such as serum amyloid A.72 Thus, HDL can be described as a “chameleon-like lipoprotein”—antiinflammatory in the basal state and proinflammatory during the acute-phase response.71 This acute-phase response, however, can also become chronic and may be a mechanism for HDL dysfunction in SLE.73
Innate Immunity in Atherosclerosis
In contrast to adaptive immunity, the components of innate immunity are present at birth and allow for immediate host defenses against pathogens. The receptors of innate immunity are known as pattern recognition receptors (PRRs); these receptors bind to preserved motifs on various pathogens termed pathogen-associated molecular patterns (PAMPs). Toll-like receptors (TLRs) are one type of PRR that respond to various PAMPs by activating their intracellular signaling pathway, leading to the upregulation of immune responsive genes.74 The ligands for TLRs include microbial ligands, a possibility that may explain some of the connections that have been postulated to exist between infectious organisms such as Chlamydia pneumoniae and the development of atherosclerosis. Endogenous ligands can also trigger TLR signaling like microbial ligands do. For example, minimally oxidized LDL interacts with TLR4 and with the scavenger receptors CD14 and CD36.75 When ox-LDL binds to the CD14 receptor on macrophages, an inhibition of phagocytosis of apoptotic cells and enhanced expression of the scavenger receptor CD36 occur, leading to increased uptake of ox-LDL. Both of these effects are thought to be proinflammatory and proatherogenic.69 Activation of TLRr7 and TLR9, resulting in the upregulation of IFN-α, has also been shown to play a major role in lupus disease activity.76 This pathway may also have implications in atherogenesis, because IFN-α plays a crucial role in premature vascular damage in SLE by altering the balance between endothelial cell apoptosis and vascular repair.77 High IFN-α levels have been associated with endothelial dysfunction in patients with SLE.78
Potential Biomarkers for Atherosclerosis in SLE
Oxidized Low-Density Lipoproteins
As already noted, the oxidation of LDLs is a triggering mechanism in the pathogenesis of atherosclerosis. In fact, high levels of circulating ox-LDL are strongly associated with documented CAD in the general population.79 There is some speculation that the increased risk of thrombotic and atherosclerotic events associated with ox-LDL may be due in part to a cross-reactivity between aCL and ox-LDL.80 Cardiolipin is a component of LDL, and anticardiolipin and anti–ox-LDL antibodies have been shown to be cross-reactive.80 Additionally, β2-GPI has been shown to bind directly and stably to ox-LDL.81 Elevations of circulating ox-LDL have been described in patients with SLE and are associated in some reports with a history of cardiovascular disease.45,82 Levels of the oxidized phospholipid 1-palmitoyl-2-arachidonyl-sn-phosphtidylcholine (ox-PAPC) have also been associated with thickened IMT on carotid ultrasound.35 ox-LDL–β2-GPI complexes are also associated with a risk of arterial thrombosis.83 Interestingly, renal manifestations of SLE have been associated with both higher levels of circulating ox-LDL82 and circulating ox-LDL/β2-GPI complexes.84
Circulating antibodies to ox-LDL (anti–ox-LDL) have also been described, although their relationship with the development and progression of atherosclerosis is unclear. Antibodies that recognize ox-LDL are generally considered to be protective against atherosclerosis in murine models,67 but one human study demonstrated a positive association between autoantibodies to ox-LDL and a history of cardiovascular disease in patients with SLE.45 Conversely, another study demonstrated that antibody titers to one phospholipid component of ox-LDL, phosphorylcholine (anti-PC antibodies), were inversely correlated with the presence of vulnerable carotid plaques in SLE.85 In two other studies, anti–ox-LDL and arterial disease were not associated.86,87 Titers of antibodies to ox-LDL have also been associated with disease activity in SLE.88
Lipoprotein(a)
In addition to oxidized LDL, lipoprotein(a) (Lp[a]) has also been implicated in the pathogenesis of atherosclerosis in both the general and SLE populations.79 Lp(a) is structurally related to LDL but also contains apolipoprotein(a), which is covalently linked to apolipoprotein B-100.89 Lp(a) has been shown to physically associate with both proinflammatory ox-LDL89 and β2-GPI.89 Circulating plasma levels of Lp(a) have been associated with CAD in the general population90 and in rheumatoid arthritis.89 Several researchers have also found elevations of Lp(a) in patients with SLE.91–93 One study reported that serum Lp(a) levels were increased in patients with lupus who also had renal disease and hypoalbuminemia and that treatment with corticosteroids reduced the elevations.93 Another group reported, however, that Lp(a) levels are not influenced by corticosteroids or disease activity.92 Lp(a) can also become oxidized (ox-Lp[a]), and levels of ox-Lp(a) and Lp(a)–β2-GPI complexes are also higher in subjects with SLE than in controls.89,94 Higher Lp(a) levels were associated on univariate analysis with increased carotid IMT in a pediatric cohort of subjects with SLE37 but were not associated with plaque in one adult cohort.95






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


