Also known as von Recklinghausen disease, NF type 1 (NF1) is one of the most common human genetic disorders. Although the primary pathologic process involves altered neuronal cell growth, NF1 may present with a number of musculoskeletal and systemic manifestations. The effect on bones, nerves, skin, and other soft tissues may lead to alterations in growth, development, and function of the upper extremity, presenting a host of challenges to the pediatric hand and upper extremity surgeon.
Etiology and Epidemiology
Historically, NF was first described in 1793 by von Tilenau, though von Recklinghausen is credited with associating the clinical manifestations of this disorder with nerve tumors.
1,2 One of the most common genetic diseases in humans, NF affects approximately 1:3,000 people and is inherited in an autosomal dominant fashion with near complete penetrance. Approximately one-half of all cases are due to sporadic mutation.
Mutations in the neurofibromin gene, localized to chromosome 17q11.2, are responsible for NF1.
11 Segmental NF typically involves multiple plexiform neurofibromas limited to a single body part without crossing the midline and devoid of the systemic clinical manifestations common to NF1.
Clinical Evaluation
Silence is golden when you can’t think of a good answer.
—Muhammad Ali
Neurofibromas refer specifically to mixed cell tumors arising from peripheral nerve sheath cells, rich in Schwann cells, fibroblasts, mast cells, and endothelial cells. The
clinical diagnosis of NF1 is made based upon a number of established clinical criteria (
Table 48-1).
1,<A onclick="if (window.scroll_to_id) { scroll_to_id(event,'R12-48'); return false; }" class=LK href="#R12-48" name=to-R12-48 xpath="/CT{06b9ee1beed5941es more specifically expressed in neuronal tissues.
These genetic abnormalities are distinct from mutations in the merlin or schwannomin gene, which have been attributed to NF type 2 (NF2) (so-called central NF).
8,9 Diagnostic criteria include the presence of eight or more nerve masses and a positive family history of a first-degree relative with NF2. It affects approximately 1:40,000 individuals and usually presents with hearing loss in young adults. While NF2 typically causes tumors of cranial nerve VIII (including the bilateral vestibular schwannomas resulting in hearing loss), meningiomas, ependymomas, and peripheral nerve tumors, musculoskeletal manifestations are uncommon. As a result, these patients rarely come under the care of the pediatric hand and upper extremity surgeon.
Histologically, neurofibromas appear as whorls of interlacing spindle-shaped cells, though they stain poorly for S-100 and Leu-7 (
Table 48-2). Technically benign, these lesions demonstrate varying morphologies, unpredictable growth patterns, and often infiltrate and involve the nerve fascicles and axons themselves.
13 Clinically, the lesions manifest themselves as solitary palpable subcutaneous nodules, often with overlying discoloration. Although rarely appral features: (1) more than six café au lait spots measuring at least 5 mm in diameter in children, (2) two or more solitary neurofibromas or one plexiform neurofibroma, (3) axillary or inguinal freckling (the so-called Crowe sign), (4) the presence of optic gliomas, (5) two or more iris hamartomas (Lisch nodules), (6) characteristic skeletal lesions (e.g., long bone pseudarthrosis), and/or (7) a firstdegree relative with NF1.
Histologically, neurofibromas appear as whorls of interlacing spindle-shaped cells, though they stain poorly for S-100 and Leu-7 (
Table 48-2). Technically benign, these lesions demonstrate varying morphologies, unpredictable growth patterns, and often infiltrate and involve the nerve fascicles and axons themselves.
13 Clinically, the lesions manifest themselves as solitary palpable subcutaneous nodules, often with overlying discoloration. Although rarely appreciated at birth, neurofibromas usually become apparent in late childhood (e.g., at the end of the first decade of life). Solitary neurofibromas account for roughly 85% of all upper limb cases.
Plexiform neurofibromas, in contrast, are characterized by diffuse, raised, hyperpigmented, cutaneous lesions with boggy or “bag-of-worms”-type consistency. Genetic mutations similar to those seen in NF1 of chromosome 17 are seen in plexiform neurofibromas; indeed, the presence of plexiform neurofibromas is almost always associated with NF1.
14 Although neurofibromas are typically nonpainful, the presence of pain or tenderness— particularly with plexiform lesions—raises the concern of malignant transformation, usually into malignant peripheral nerve sheath tumors (MPNST).
15, Figure 48-1). When left untreated, the condition may result in bowing deformities of the limb (particularly the leg). Given the abnormal biology of these lesions, it is common to see persist941965993857848f162b18c0277f5bd9542aaccd047c370a32ebc50f9af14c097802b99ae17586658032}/ID(R17-48)” title=”17″ onmouseover=”window.status=this.title; return true;” onmouseout=”window.status=”; return true;”>17 and
18 Indeed, owing to the genetic basis of the disease, patients with NF are at higher risk for other malignancies as well, including melanoma, rhabdomyosarcoma, astrocytomas, and hematologic malignancies.
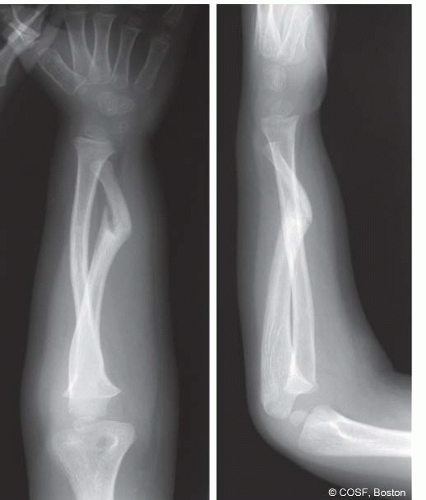
In addition to these classic and diagnostic clinical manifestations, NF may cause bony lesions resulting in presentation to a pediatric hand and upper extremity surgeon. Perhaps the most common and severe orthopaedic issue related to NF is pseudarthrosis of the long bones, most commonly the tibia, fibula, ulna, and clavicle.
19 While only 5% to 10% of patients with NF will demonstrate bony pseudarthroses, up to 75% with pseudarthroses will have NF1.
13,20,21 These patients typically present with deformity of the affected limb, often with purported
history of prior fracture nonunion. While the exact pathophysiology is unknown, these bone lesions are characterized radiographically by nonunion sites devoid of typical periosteal reaction and callus formation, with dysplasia, sclerosis, and narrowing of the adjacent bony segments (see
Coach’s Corner) (<A onclick="if (window.scroll_to_id) { scroll_to_id(event,'F1-48'); return false; }" title="Figure 48-1" class=LK href="#F1-48" name=to-F1-48 xpath="/CT{06b9ee1beed5941965993857848f162b18c0277f5bd9542aaccd047c370a32ebc50f9af14c097802b99ae17586658032}/ID(F1-48)" cellspacing="0" o0? (
Table 48.2 Features distinguishing schwannoma from neurofibromas |
|---|
|