Chapter 13 Neural-Immune Interactions
Principles and Relevance to SLE
The immune and central nervous systems are the body’s primary tools for interfacing with constant environmental perturbations that threaten homeostasis. Chemical, antigenic, or infectious agents recognized by the immune system and psychological or physical stimuli recognized by the central nervous system (CNS) often activate similar transducing pathways to translate perturbing signals into stabilizing responses. Numerous studies (human and animal model) also provide evidence for bidirectional communication between the CNS and the immune system. Cytokines produced by cells of the immune system stimulate the CNS, leading to “sickness behavior” following an infection—characterized by loss of appetite, decreased mobility, loss of libido, withdrawal from social interaction, depressed mood, increased somnolence, and fever—whereas hormones and proteins generated by the CNS modulate immunity to influence the course of immune-related disease (Figure 13-1).1 Disruptions in communication between these systems can increase susceptibility to and severity of a variety of diseases, including autoimmune and inflammatory conditions such as systemic lupus erythematosus (SLE).2 This chapter outlines the general principles of communication between the CNS and immune system and how this interaction could play a significant role in SLE disease outcome. In addition, it defines the afferent and efferent limbs of CNS regulation of immunity, the contribution of specific immune cell populations and their actions in initiating autoimmune/inflammatory conditions, and evidence from studies with humans and using animal models that demonstrate how this interaction operates in SLE.
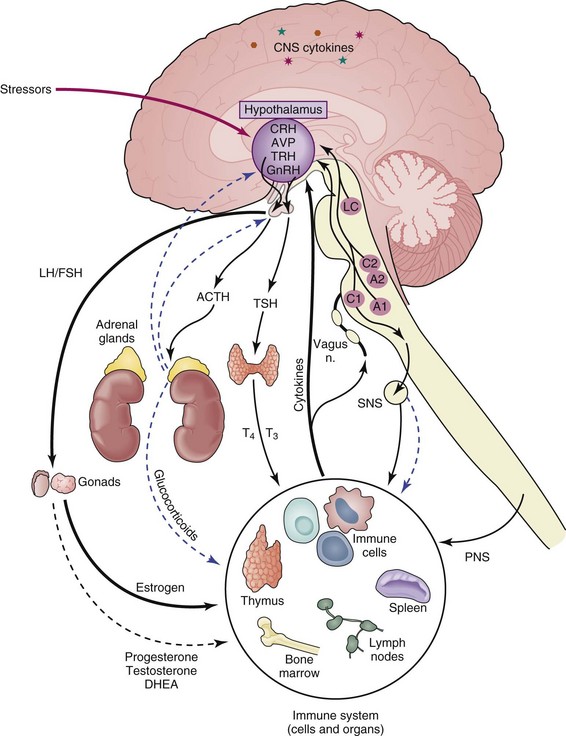
Communication between the CNS and the immune system impacts physiologic processes at multiple levels (local, regional, systemic), and interruptions at any point in this dialogue could disrupt homeostasis and lead to disease. The strength of an immune response and of inflammation when a foreign stimulus is encountered depends not only on the nature, potency, dose, route, and duration of exposure but also on the contribution of CNS influences—including under conditions of stress.3 This fact has important implications with the use of pharmacologic agents because drugs aimed at ameliorating autoimmune/inflammatory disease could alter the course of disease if they modify activity of the CNS or be ineffective when administered while the individual is stressed, because hormones generated during the stress response have a profound effect on immune responses and are likely to affect disease outcome.
The Immune System
Activation of the immune system is important for preventing disease when a pathogen is introduced; however, if uncontrolled, immunopathology can result in autoimmune/inflammatory conditions. Responses generated by the immune system are generally divided into two groups: innate and adaptive. Innate immunity provides early—minutes to hours—immunologic events and an initial defense against pathogens and also supplies signals (cytokines, chemokines, costimulatory molecules, etc.) necessary to stimulate adaptive immunity.4,5 Several different cell types are involved in innate immunity, including granulocytes, monocytes, dendritic cells (DCs), and natural killer (NK) cells. Granulocytes (basophils, eosinophils, and neutrophils) are found throughout the body and are among the first immune cells recruited after an injury.6,7 Monocytes originate in the bone marrow and are a population of antigen-presenting cells (APCs) with phagocytic properties that can produce cytokines and chemokines to attract other immune cells to initiate an inflammatory response.8,9 DCs are a more potent APC population that recognizes pathogens using receptors for pattern-associated molecular patterns (PAMPs) to drive strong immune responses by producing cytokines and expressing molecules on their cell surfaces that stimulates other immune cells.10,11 NK cells have cytotoxic function and kill by producing perforin and granzyme, which break up the plasma membrane of a target cell or induce apoptosis using death receptors, such as Fas ligand (FasL) and tumor necrosis factor (TNF)–related apoptosis–inducing ligand (TRAIL).7,12
Adaptive immunity provides long-term protection against such pathogens as viruses, parasites, and tumor cells. Although the time required to initiate adaptive immune responses is more extensive (days to weeks), a second exposure to the pathogen is eliminated much more rapidly.13,14 The majority of autoimmune/inflammatory conditions, such as SLE, are mediated by overactive or uncontrolled adaptive immune responses; therefore, controlling adaptive immunity is critical. Cells of adaptive immune responses consist primarily of B and T lymphocytes but require the efforts of innate immune cells to drive their responses. B cells are antibody-producing cells that develop in bone marrow and form germinal centers—sites of B-cell proliferation—upon activation to generate humoral immune responses, and these cells’ lack of controlled activity is a key component in development of SLE.15,16 Antibodies are important for neutralizing pathogens but can also stimulate other immune cell activity, including complement-mediated immune responses. T cells, also an important population in adaptive immunity, generate cellular immunity, including CD8+ T cells, which are cytotoxic to target cells (similar to NK cells) and assist in the promotion of other immune responses. The various immune cell populations work together to provide the host with an efficient system for eliminating pathogens and ameliorating disease, and additional information on the involvement of specific populations of innate and adaptive immune cells in SLE is discussed elsewhere in this text.
Central Nervous System Regulation of Immunity
The Neuroendocrine System
The neuroendocrine system comprises the hypothalamus, the pituitary, and glands involved in release of their respective hormones, that is, the adrenal glands and the gonads. The HPA axis exerts its effects primarily through release of glucocorticoids and mineralocorticoids17,18 by the cortex of the adrenal glands. The neuroendocrine stress response generated through the HPA axis is activated by stressful stimuli and is a powerful regulator of immune responses. The HPG axis exerts its effects on immunity through the sex hormones estrogen, progesterone, and testosterone.19,20 These hormones bind to a family of related nuclear hormone receptors. Activity of the HPG axis fluctuates throughout the life cycle, whereas activity of the HPA axis tends to be more consistent. Together these systems can affect immunity to, susceptibility to, and the course of autoimmune/inflammatory diseases. Temporal and quantitative changes in these systems and their effects on immunity are discussed in detail later.
The Stress Response
The stress response can be defined as the brain’s physiologic and behavioral reaction to psychological or physical stressors. The hypothalamus responds to internal and external stimuli by synthesizing the neuropeptide corticotropin-releasing hormone (CRH) from cells of the paraventricular nucleus (PVN)—neurons in the hypothalamus that project to the sympathetic brainstem nuclei, parasympathetic brainstem preganglionic neurons, and spinal cord.21 CRH secreted into the rich hypophyseal portal blood supply stimulates the anterior pituitary gland to secrete adrenocorticotropin hormone (ACTH), which in turn stimulates the adrenal glands to synthesize and secrete glucocorticoids.22 Hypothalamic CRH secretion is held under tight regulatory control by several positive and negative neurotransmitter systems to regulate glucocorticoid release from the adrenal glands. The noradrenergic, serotonergic, and dopaminergic systems upregulate CRH via α1-adrenergic receptors,23 serotonin (5-hydroxytryptamine [5-HT]) receptors,24 and dopamine (D1) receptors,25 respectively, whereas opiates, gamma-aminobutyric acid (GABA)/benzodiazepine, and glucocorticoid feedback suppress CRH production via opiate receptors,26 GABAergic receptors,27 and glucocorticoid receptors,28 respectively.29,30
In addition to its neuroendocrine effects via the pituitary gland, CRH also acts centrally within the brain as a neuropeptide to induce a set of behaviors that are characterized by cautious avoidance, vigilance, enhanced attention, and suppression of vegetative functions, such as feeding and reproduction,31,32 which make up the classic “fight or flight” pattern of behavior.23,33 Many of these effects are mediated through the hypothalamus and neurotransmitter systems, such as the brainstem-noradrenergic system and sympathetic nervous system.34,35 The hypothalamic CRH system also communicates with noradrenergic pathways during the stress response via anatomic connections between the hypothalamus and noradrenergic centers in the brainstem.36 In turn, the brainstem-noradrenergic system sends signals to the periphery via the sympathetic nerves. Through such connections, the physiologic components of the stress response (i.e., increased heart rate, muscle tone, and sweating) are coordinated with behavioral responses to form the generalized stress response. Many studies suggest that modulation of immune responses by both the sympathetic and neuroendocrine systems are an important physiologic component of the stress response.37–42
Sex Hormones
Another group of steroid hormones whose production is controlled by the neuroendocrine system and that are shown to be important in autoimmune/inflammatory conditions are sex hormones, and their involvement in SLE is discussed in more detail elsewhere in this text. Both males and females produce the different sex hormones—estrogen, progesterone, testosterone—but with varying concentrations. These hormones are critical in the development of secondary sexual characteristics and during pregnancy but also play a role in modifying immune responses. The hypothalamus responds to rhythmic signals to produce gonadotropin-releasing hormone (GnRH).43,44 GnRH acts to stimulate pituitary gland cells to produce luteinizing hormone (LH) and follicle-stimulating hormone (FSH), which enter the bloodstream and interact with gonadal cells (testes, ovaries) that activate gametogenesis and produce sex hormones, such as testosterone by interstitial cells and estrogen and progesterone by granulosa cells.45 Production of sex hormones in females is especially sensitive to rhythmic changes throughout the reproductive cycle.46
Interactions between the HPA and HPG Axes
There is an important interplay between the HPA and HPG axes. In addition to regulating its own activity, the HPA axis is able to modify activity of the HPG axis, and vice versa. Psychological or physical stressors that generate elevations of glucocorticoids for extended periods or other events leading to overactivity of the HPA axis can initiate a negative feedback loop by acting on the hypothalamus and limit further production of glucocorticoids. Stressors that cause classic “sickness behavior” can also reduce the libido and limit production of sex hormones by suppressing HPG axis activity.47 Conversely, the HPG axis is able to regulate activities of the HPA axis, such as elevated concentrations of sex hormones limiting glucocorticoid release by adrenal glands.48 Co-regulation between the HPA and HPG axes could, therefore, impact activation of the immune system and potency of immune responses following a trigger.
Molecular Mechanisms of Steroid Hormone Actions
Receptors for glucocorticoids and other steroid hormones are members of the nuclear receptor superfamily and are structurally related. They include glucocorticoid receptor (GR), which that binds corticosterone and dexamethasone; mineralocorticoid receptor (MR), which binds corticosterone and aldosterone; androgen receptor (AR), which binds testosterone and its derivatives; estrogen receptor, which binds estradiol (ER); progesterone receptor (PR), which binds progestins; thyroid hormone receptor, which binds thyroxine; and retinoic acid receptors, which bind all-trans retinoic acid. Structurally, these receptors are made up of three functional regions: (1) a C-terminal hormone–binding region, (2) a DNA-binding region, and (3) an N-terminal immunogenic region involved in transactivation.49,50 The unbound receptor located in the cytosol is folded and inactive, bound to a 90-kilodalton (kDa) heat shock protein51 (HSP90) and immunophilins (Figure 13-2). When the ligand (hormone) binds to its receptor, hsp90 is displaced, resulting in a conformational change in the receptor that allows the active ligand-receptor complex to displace to the nucleus and bind to hormone receptor–binding elements (HREs) on DNA as either a homodimer or heterodimer. For example, although GR generally binds to GR DNA-binding elements52 as homodimers, it is also possible for GRs and MRs to form heterodimers.53,54 These different mechanisms of binding to DNA response elements confer additional specificity of action to the steroid hormone receptors. The hormone receptor complex then translocates to the nucleus and acts as a transcription factor, either suppressing or stimulating DNA gene transcription. In addition, the GRs and other steroid hormone receptors interact with more than 200 nuclear cofactors. The recruitment of either coactivators or co-repressor complexes is involved in transcriptional regulation and can determine whether or not a gene is transcribed.55 Other accessory proteins, such as (histone deacetylase 6) HDAC6, also contribute to transcriptional regulation,56 and steroid hormone receptors can interact with other transcription factors, such as NF-κB and (activator protein 1) AP-1, to inhibit their activity in immune and other cells.57,58
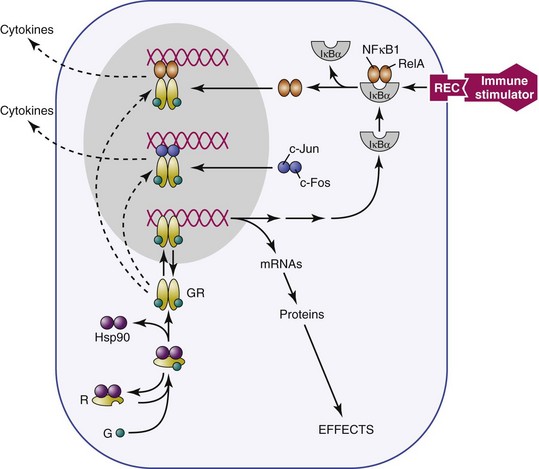
Further specificity of action of these receptors is conferred by tissue distribution within tissues. This process is well-documented for the relationship between GRs and MRs. The primary glucocorticoid receptor in immune cells is GR, which is consistent with the physiologic role of glucocorticoid regulation of the immune system by stress levels of these hormones1,59,60; however, an additional level of specificity is conferred by tissue distribution of the corticosterone-metabolizing enzyme 11β-hydroxysteroid, which metabolizes corticosterone but not aldosterone. Thus, where 11β-hydroxysteroid is present (e.g., kidney), the primary ligand that is available for binding to MRs is aldosterone rather than corticosterone,52 whereas where the enzyme is not present (e.g., brain), the primary ligand for MRs is corticosterone. MRs in the brain play a role in regulation of basal HPA function, such as circadian rhythm.
Impact of Neuroendocrine Factors on Immunity
Several neuroendocrine factors, including steroid hormones, have been shown to alter immunity and impact immune-related disease outcome. This effect was demonstrated dramatically when it was shown that hormonal fluctuations could influence the size of lymphoid organs, such as experiments using restraint and psychological stressors that activate the HPA axis and led to shrinkage of the thymus and other lymphoid tissues.61 In addition, many of the autoimmune/inflammatory conditions exhibit differences in incidences in males and females (being up to tenfold higher in women), suggesting a role for sex hormones in immune-mediated disease.
Glucocorticoid Modulation of the Immune System
Glucocorticoids are able to modulate immune cell function by acting through intracellular GRs in immune cells. The overall functional effect of glucocorticoids on the immune response depends on the preparation, dose (whether pharmacologic or physiologic), and temporal sequence of glucocorticoid exposure in relation to antigenic or proinflammatory challenge and has profound effects at molecular, cellular, and whole-organ levels.62–64 Exposure to stress levels of glucocorticoids results in rapid involution of the thymus as a result of glucocorticoid-induced thymocyte apoptosis, and glucocorticoids can regulate immune responses by inducing apoptosis in proliferating lymphocytes.49 There is evidence to suggest that such glucocorticoid-regulated apoptosis could take place within the thymus through induction of an intrathymic glucocorticoid system because the enzymatic machinery for glucocorticoid synthesis is present within the thymus.65 Glucocorticoids also orchestrate redistribution of circulating white blood cells with neutrophilic leukocytosis, eosinopenia, monocytopenia, and altered ratios of T-lymphocyte subtypes—resulting in decreased peripheral blood CD4+ cells and increased CD8+ cells—as well as decreased infiltration of neutrophils and monocytes into tissues.66
Glucocorticoids have effects on both innate and adaptive immune cell populations, including granulocytes, NK cells, monocytes, DCs, and B and T lymphocytes.67 Mice treated with glucocorticoids show a reduction in the number of splenic NK cells, and remaining NK cells exhibit reduced cytolytic activity.68 Glucocorticoids inhibit cytokine release and other activity of eosinophils in asthma69,70 and of neutrophils in chronic obstructive pulmonary disease (COPD),71,72 and inhaled glucocorticoids prevent histamine release by basophils in allergic disease.73 Monocytes and neutrophils are thought to be primary targets of glucocorticoid actions in diminishing contact hypersensitivity reactions, as evidenced by repression of monocyte production of cytokines and chemokines.74 Glucocorticoids have also been shown to reduce DC production of interleukin-12 (IL-12), limit upregulation of co-stimulatory molecules expressed by mature DCs to reduce recognition of antigen, and strongly reduce allostimulatory capacity.74,75 However, the suppressive effect was not observed with DCs previously activated by lipopolysaccharide (LPS), indicating that the influence of glucocorticoids depends on stage of DC maturation.76 In a study of children with asthma, glucocorticoids were shown to decrease expression of intracellular adhesion molecule 1 (ICAM-1) and L-selectin, leading to an inhibition of the ability of immune cells to migrate to inflammatory sites.77
In addition to effects on innate immune cells, extensive studies have shown immunomodulatory consequences of glucocorticoids on adaptive immunity. Glucocorticoids suppress differentiation and maturation of T cells as well as altering the function of T-cell subtypes, such as cytolytic (CD8+) and helper (CD4+) T cells. In addition, several studies have reported that glucocorticoids suppress mitogen- and antigen-stimulated T-cell proliferation.78 This is thought to be most critical in T-helper (Th) cell populations, which are skewed from a Th1 toward a Th2 or other T-helper cell responses in the presence of glucocorticoids, with inhibition of TNF-α, IL-2, IL-6, IL-12, and interferon gamma (IFN-γ) production and increases in IL-10, IL-4, and IL-13 production.79–81 The impact of glucocorticoids on B-cell proliferation is variable, depending on the stimulus and dose of glucocorticoids used and age-dependent expression of GRs.82,83 In general, B-cell proliferation is suppressed by glucocorticoids to a lesser extent than T-cell proliferation, but suppression of Th2 subsets that assist in antibody production could inhibit B-cell activity. Although the overall effects of glucocorticoids on immune responses at the cellular level are immunosuppressive, this effect is attained through suppression of many stimulatory components of the immune cascade and stimulation of some immunosuppressive or antiinflammatory elements. The relatively greater sensitivity to glucocorticoid suppression of components of cellular versus humoral immunity tends to shift immune responses from a cellular to a humoral pattern,1 which is important in SLE.
Effects of Sex Hormones on Immunity
Immune suppression or modulation of immunity by sex hormones has been reported in many diseases.84–87 In female mice, surgical removal of ovaries (oophorectomy, essentially eliminating available estrogen and progesterone) followed by hormone treatment, such as estrogen, has been used to show their effects on immune responses. In male mice that have undergone castration (orchidectomy) and been given estrogen, increases in susceptibility to autoimmune/inflammatory disease to levels that are similar to those in females have been reported. Sex hormones can have direct effects on immune cells because they express receptors for estrogen (ERs), progesterone (PRs), and testosterone (ARs) but may also modify immunity through indirect effects on the HPA or HPG axis.88,89 In the female genital tract, the number of uterine NK cells changes during the reproductive cycle and with pregnancy,90 and progesterone receptor (PR)−/− mice do not have uterine NK cells.91 In addition, women are more susceptible to a variety of infections during pregnancy, including the bacterium Listeria monocytogenes, which poses a significant health problem.92 Taken together, these findings indicate a role of sex hormones in the regulation of immunity.
Although the impact is not as dramatic as has been shown with glucocorticoids, sex hormones are able to modify both innate and adaptive immune cell populations. Estrogens can prevent production of free radicals93 and adhesion to endothelial cells by neutrophils94; in ovariectomy experiments using mice, an infiltration of neutrophils was found in the endometrium95,96; and monocyte populations are also responsive to sex hormones. Bacterial lipopolysaccharide–activated splenic macrophages treated with estrogen have reduced production of proinflammatory cytokines.97 Testosterone induces Fas ligand (FasL)–dependent apoptosis in bone marrow–derived macrophages,98 and progesterone increases expression of FasL, inhibits TNF-α production in uterine macrophages,99 and decreases cytokine (IL-12, IL-1β) production by monocytes from varicella-zoster virus (VZV)–stimulated peripheral blood mononuclear cells (PBMCs) from healthy subjects.100 Progesterone has also been reported to inhibit DC phagocytosis of Candida albicans,101 to contribute to susceptibility to human immunodeficiency virus (HIV) in women by increasing the number of Langerhans cells available for HIV infection,102 and to increase susceptibility to Chlamydia infection in rats as a result of increased bacterial infiltrates in the uterine epithelium and vaginal secretions of these animals.103 In addition, estrogen has been shown to affect DC function in the development of experimental autoimmune encephalomyelitis (EAE)—the mouse model for the autoimmune disease multiple sclerosis.104,105
Sex hormones also have a profound influence on adaptive immune responses. The two forms of estrogen receptor, ERα and ERβ,106 have been identified in lymphocytes,107 and the presence of estrogen increases the severity of autoimmune diseases driven by B and T cells in mice and humans. Progesterone inhibits T-cell proliferation,108 whereas estrogen at elevated concentrations (achieved during pregnancy) is thought to be important in immune suppression during pregnancy, possibly by increasing T-regulatory cell (Treg) populations to prevent fetal rejection.109,110 In addition, estradiol has been shown to increase antibody production by B cells and has been implicated in the improper regulation of B-cell development.15,114 The ability of estrogen to aggravate SLE symptoms is related at least in part to dose. In one study, administration of hormone replacement therapy (Premarin 0.625 mg (CLB) conjugated estrogen) to women with SLE increased their risk for mild-to-moderate flares of disease.111 In another study, however, administration of lower doses of estradiol-containing oral contraceptives (30 to 35 µg (CLB) estradiol, plus methindrone), compared to placebo, to women with stable SLE (and no history of clotting) was not associated with increased flare rates.112
Autonomic and Peripheral Nervous System Regulation of Immunity
The CNS can also utilize the autonomic and peripheral nervous systems to regulate immune responses and influence development of autoimmune/inflammatory conditions.3 The sympathetic nervous system releases neurotransmitters, such as norepinephrine, and connects CNS adrenergic brainstem regions to lymphoid organs.113,114 The parasympathetic nervous system modulates immune responses through efferent and afferent fibers of the vagus nerve. The peripheral nervous system regulates immunity through release of neuropeptides from sensory peripheral nerves involved in pain, touch, and temperature perception. Interplay between these systems provide the feedback loop that optimizes immune responses by amplifying immunity to clear a pathogen and then dampening the response to prevent overactivation of the immune response that would lead to autoimmune/inflammatory conditions, such as SLE.
Sympathetic Nervous System Effects on Inflammation and the Immune System
The sympathetic nervous system serves an important role in regional regulation of immunity.115,116 Many lymphoid organs—including spleen, thymus, and lymph nodes—are richly innervated by sympathetic nerves.117 A number of studies, including denervation and ablation studies, indicate that these anatomic connections as well as the neurotransmitters of the sympathetic nervous system play an important physiologic role in inflammatory responses.115 The sympathetic nervous system mediates its effects through release of norepinephrine from sympathetic nerve fibers and epinephrine released from the adrenal medulla. Both norepinephrine and epinephrine exert their actions through adrenergic receptors. The beta-2 adrenergic receptor (β2AR), a G-protein–coupled receptor, is the main receptor found on lymphocytes.118,119 Adrenergic influences on immune cells potently inhibit Th1 cytokine production and thereby suppress cell-mediated immune responses. Norepinephrine and epinephrine also stimulate production of Th2 cytokines, such as IL-10, that could drive immune cell activity in SLE.120
Evidence that the sympathetic nervous system affects the exudation component of peripheral inflammation is provided by sympathetic ablation studies using 6-OH dopamine (6-OHDA)121 or sympathetic ganglionic blockers such as chlorisondamine,122 or noradrenergic antagonists and agonists.123 Noradrenergic denervation studies using rodent models have shown differential effects on inflammation depending on the location of denervation. Thus, denervation of the noradrenergic fibers of lymph nodes124 is associated with exacerbation of inflammation, but systemic sympathectomy or denervation of joints is associated with decreased severity of inflammation.125,126 Treatment of neonatal rats with 6-OHDA, which interrupts both central and peripheral noradrenergic systems, is associated with exacerbation of experimental allergic encephalomyelitis.127 Pharmacologic studies show decreased inflammation in experimental arthritis with beta-blockade128 and decreased severity of experimental allergic encephalomyelitis with β-adrenergic agonists.127

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


