Chapter 48 Myopathic Disorders
Clinical and Diagnostic Approach to the Patient With a Suspected Myopathy
Disease History
A comprehensive past medical history and surgical history should be obtained in the patient suspected of having a myopathy (Box 48-1). The most common presenting complaints from patients with myopathic disorders include weakness, fatigue, and lack of endurance. Parents of children with suspected myopathic disorders often report a history of infantile floppiness or hypotonia, delay in motor milestones, feeding and respiratory difficulties, abnormal gait characteristics, frequent falls, difficulty ascending stairs or arising from the floor, muscle cramps, or stiffness. Teenagers or adults with later-onset disorders typically present with chief complaints of strength loss or decreasing endurance, falls, difficulty ascending stairs, exercise intolerance, episodic weakness, muscle cramps, focal wasting of muscle groups, and breathing difficulties.
BOX 48-1 Clinical History in Myopathic Disorders
Family History
In autosomal recessive conditions, only one generation is usually affected, with equal incidence in males and females. One fourth of offspring are typically clinically affected. Parents in earlier generations might be normal, and the parents of affected children are presumptive heterozygote carriers of the condition. In many instances of autosomal recessive inheritance, no other members of the nuclear family unit are affected. This makes the confirmation of inheritance pattern difficult unless a molecular genetic marker is present or protein abnormality is confirmed by immunohistochemistry techniques.28
Examination of the affected relatives is important. The medical records and diagnostic evaluations of affected family members should be reviewed and the diagnosis confirmed if possible. Did other members have disease onset at the same time and similar progression? In some instances, the examination of a parent can help establish the diagnosis in an affected infant or child, as is frequently the case in myotonic muscular dystrophy type 1 (DM1). In this disorder, genetic anticipation with abnormal CTG trinucleotide expansion of unstable DNA results in progressively earlier onset and increasing severity of the disease in successive generations.2,117 In the case of the hereditary myopathies, a definitive molecular genetic or pathologic diagnosis can often be found in a sibling or close relative that can assist in the establishment of the diagnosis in the patient. The clinician can often establish the diagnosis in a child or adult based on clinical examination and easily obtained laboratory data, creatine kinase (CK) level, or molecular genetic testing. This is often sufficient to avoid invasive testing such as muscle biopsy.
Anesthetic History
A thorough anesthetic history should be obtained. Malignant hyperthermia (MH) is associated with primary familial malignant hyperthermia syndrome (MHS), which is most commonly caused by a gene defect in the ryanodine receptor gene at 19q13 (MHS1). This has also been linked to central core congenital myopathy. Six subtypes of MHS exist, and MH has also been seen in carnitine palmitoyltransferase II (CPT2) deficiency. Inhalational anesthetic agents (with or without succinylcholine) have also been implicated as a cause of acute rhabdomyolysis and hyperkalemia that can resemble MH. This has been most commonly reported in those with Duchenne muscular dystrophy (DMD) and Becker muscular dystrophy (BMD).49 Rare reports of MH exist in myotonic dystrophy, myotonia congenita, and Schwartz-Jampel syndrome.
Physical Examination
Physical examination findings typically help focus the remainder of the diagnostic evaluation, such as electrodiagnosis, molecular genetic testing, and histopathologic analysis of biopsy specimens. All diagnostic information must be interpreted within the context of relevant clinical information. In many instances, a precise molecular genetic diagnosis might not be possible. The diagnostic workup should determine the most likely clinical syndrome, however, and allow the clinician to provide the patient and family with accurate prognostic information and guidance. Table 48-1 shows the typical time of onset of the physical findings observed in the most common myopathic diseases.
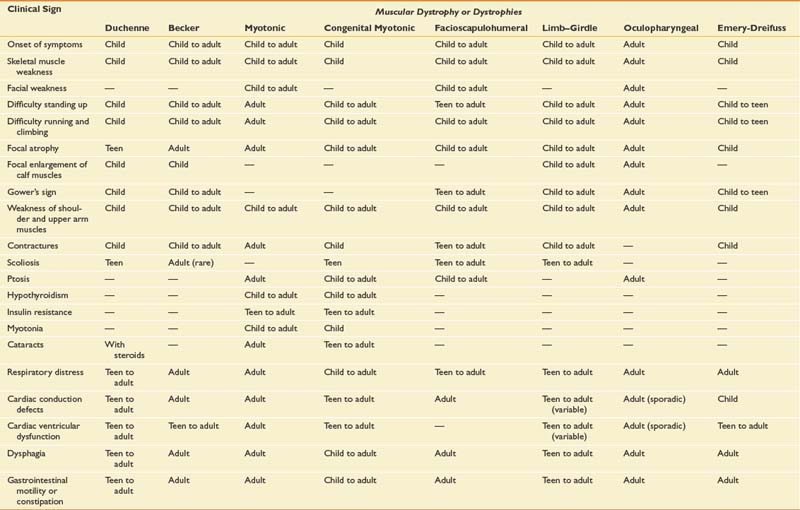
Specific aspects of the physical examination include simple inspection for the presence of focal or diffuse muscle wasting. Focal enlargement of muscles or “pseudohypertrophy” (Figure 48-1) can be seen in such dystrophic myopathies as DMD and BMD. The increase in calf circumference in DMD and BMD is caused by an increase in fatty connective tissue rather than true muscle fiber hypertrophy.13,15,33 Other neuromuscular disorders can show calf pseudohypertrophy, such as childhood-onset acid maltase deficiency. Focal atrophy of particular muscle groups can provide diagnostic clues to specific myopathic disorders, such as with Emery-Dreifuss muscular dystrophy type 1 (EMD1), facioscapulohumeral muscular dystrophy (FSHD), and selected limb–girdle muscular dystrophy (LGMD) subtypes.
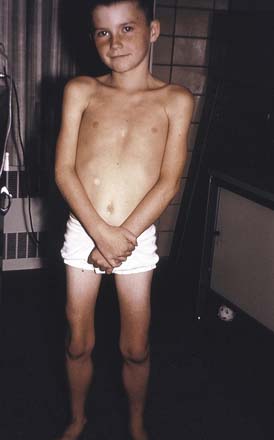
FIGURE 48-1 Boy with Duchenne muscular dystrophy demonstrating pseudohypertrophy of the deltoid and calf musculature.
(Courtesy Randall L. Braddom, M.D.)
A thorough general physical examination of cardiac, pulmonary, and gastrointestinal systems should be performed on all patients suspected of having a neuromuscular disease. Hepatomegaly can be seen in such metabolic myopathies such as acid maltase deficiency (type 2 glycogenosis) and type 3 and 4 glycogenosis. The skin should be evaluated for characteristic skin rashes and nail bed capillary changes if an inflammatory myopathy such as dermatomyositis is suspected. Craniofacial changes and dental malocclusion are common in congenital myotonic muscular dystrophy (MMD), congenital myopathies, and congenital muscular dystrophy (CMD). A neurologic examination should include a thorough evaluation of cranial nerve function, muscle tone, muscle strength, sensory and cerebellar function, muscle stretch reflexes, and assessment for the presence of percussion and/or grip myotonia in situations where a myotonic syndrome is suspected (Figure 48-2). Musculoskeletal examination often shows the presence of limb contractures and spinal deformity. Congenital contractures are common in CMD. In many dystrophic myopathies, flexion contractures can develop over time in the hips and knees, whereas equinovarus contractures can develop in the ankles. Focal elbow flexion contractures might be present in patients with EMD1 while they are still ambulatory, and they occur in DMD subsequent to transition to the wheelchair. Significant intellectual impairment can occur in some myopathic disorders, such as congenital MMD, Fukuyama CMD, selected cases with mitochondrial encephalomyopathies, and a small proportion of DMD cases.
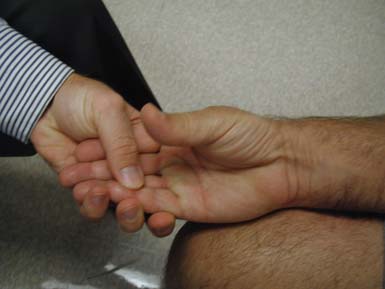
FIGURE 48-2 Percussion myotonia of the thenar eminence in a patient with myotonic muscular dystrophy.
Careful assessment of scapular winging, scapular stabilization, and scapular rotation is very helpful in the assessment of patients with FSHD and some LGMD subtypes (Figure 48-3). The scapula is stabilized for overhead abduction by the trapezius, rhomboids, and serratus anterior. Shoulder abduction to 180 degrees requires strong supraspinatus and deltoid muscles, in addition to strong scapular stabilizers.
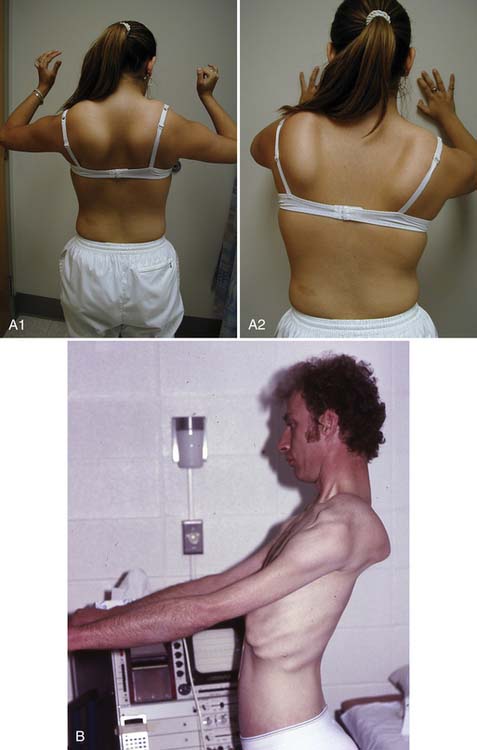
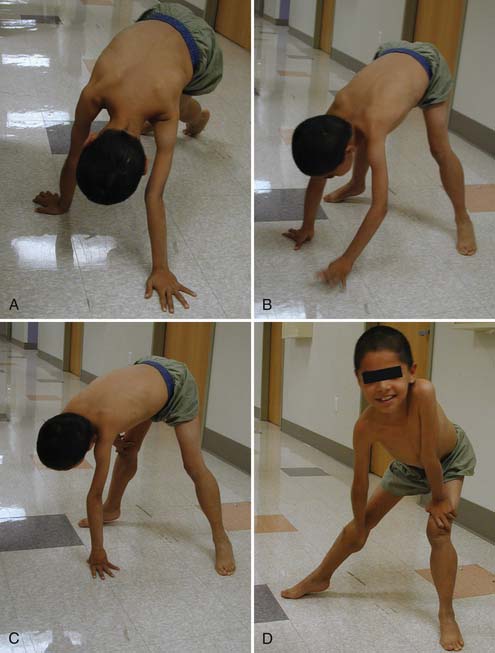
Patients with proximal weakness involving the pelvic girdle muscles often rise from the floor using the classic “Gower’s sign.” The patient usually assumes a four-point stance on knees and hands, brings the knees into extension while leaning forward on the upper limbs, substitutes for hip extension weakness by pushing off the knees with the upper limbs, and sequentially moves the upper limbs up the thigh until an upright stance with full hip extension is achieved (Figure 48-4). A Gower’s sign is not specific to any myopathic condition but can be seen in DMD, many forms of LGMD including childhood-onset autosomal recessive LGMD (LGMD2), CMD, and congenital myopathy.
Serum Laboratory Studies
Most myopathic conditions show significant elevations in transaminases, aldolase, and CK.40 The CK enzyme catalyzes the release of high-energy phosphates from creatine phosphate. It is present mainly in muscle and leaks into the serum in large amounts in any disorder involving muscle fiber injury. The MM (muscle myomesin) fraction is specific to skeletal muscle. The CK value is significantly elevated in early stages of DMD and BMD, with values up to 50 to 100 times normal. A normal CK value essentially excludes the diagnosis of DMD and BMD. Overlap in CK values occurs between DMD and BMD. Other forms of muscular dystrophy such as EMD1, LGMD, FSHD, and CMD can show moderate elevations in CK. In CMD, however, the CK value is extremely variable, ranging from normal to a fairly marked elevation. Neuromuscular disease severity is not closely associated with CK values. In all dystrophic myopathies, the CK values tend to decrease over time because the patient has progressive loss of muscle fibers as a result of irreversible cell death. For example, a 3-year-old with DMD might have a CK value of 25,000, whereas a 10-year-old with DMD could show a CK value of only 2000. This is because as muscle cells die, there are fewer of them left to leak CK into the serum.
Other conditions with significant elevations in CK include polymyositis, dermatomyositis, acute rhabdomyolysis, and familial MH. The serum CK is likely to be normal or only mildly elevated in many of the congenital structural myopathies, such as central core disease, nemaline rod myopathy, and fiber-type disproportion syndrome. In a child with muscle weakness, a normal CK does not exclude a myopathy. However, a severely elevated CK is highly suggestive of a dystrophic myopathy or inflammatory myopathy.
Lactate and pyruvate levels are useful in the evaluation of possible metabolic myopathy. The presence of a lactic acidosis can be seen in such mitochondrial encephalomyopathies as Kearns-Sayre syndrome, myoclonic epilepsy and ragged red fibers (MERRF), and mitochondrial encephalomyopathy with lactic acidosis and strokelike episodes (MELAS). Whenever clinical evidence suggests a disorder of oxidative metabolism, blood lactate and pyruvate levels should be obtained. Arterial lactate values are a more reliable guide. Stable or reduced levels of lactate and pyruvate with concomitant increases in ammonia with ischemic or nonischemic forearm exercise testing suggest a mitochondrial dysfunction. In a setting of lactic acidemia, the lactate/pyruvate ratio can aid in the differential diagnosis. Children with suspected mitochondrial encephalomyopathy should be evaluated with cerebrospinal fluid (CSF) lactate and pyruvate levels because these values are less subject to flux than are either venous or arterial values. The ischemic forearm test initially used by McArdle and the nonischemic forearm exercise test are widely used means of assessing muscle and aerobic metabolism in older, more cooperative patients.59,69
Electrodiagnostic Studies
The increased application of diagnostic molecular genetic testing in hereditary myopathies has led to a reduction in the reliance on needle electromyography (EMG) in the diagnostic workup of these patients. Needle EMG is still used to help select an appropriate muscle for biopsy. Most electromyographers limit their needle EMG study to one side of the body so that the contralateral side can be biopsied, thus avoiding potential interference from the needle trauma or minor hemorrhage.
Muscle Biopsy Evaluation
Muscle Biopsy Technique
The two techniques for obtaining a muscle biopsy specimen include traditional open biopsy and needle biopsy.39,92 Either technique can be performed under local anesthesia, although most clinicians in the United States use general anesthesia for open biopsies and local anesthesia for needle biopsies. There can be some disruption in the architecture of the tissue with needle biopsy technique, which can affect the histologic examination and electron microscopy. Immunocytochemistry analyses, such as Western blot and metabolic studies, do not require strict preservation of the muscle cellular architecture.
Histology and Histochemistry
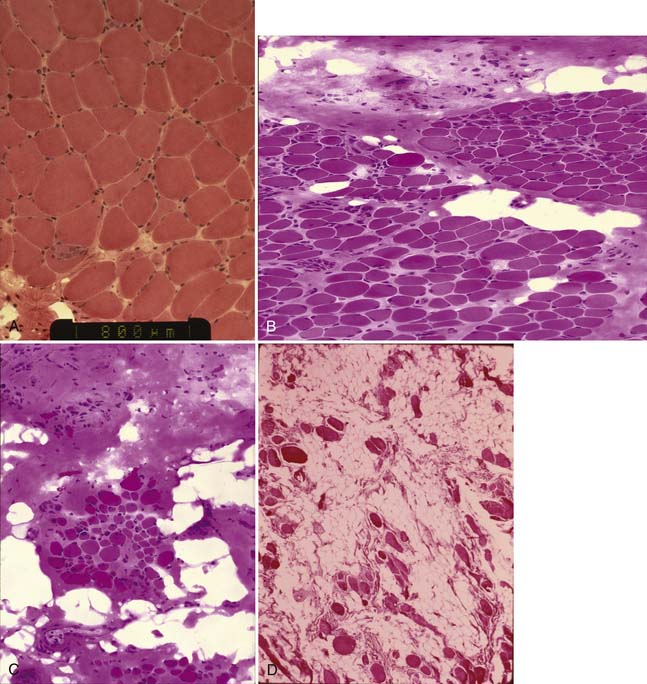
The histopathologic study is likely to provide information about whether the basic disease process is primarily a myopathy or a neurogenic process. In some instances, a specific diagnosis can be made (such as dystrophic myopathy or inflammatory myopathy). When analyzing paraffin sections, basic pathologic reactions of muscle can include fiber necrosis, central nuclei indicative of regeneration, abnormalities of muscle fiber diameter (atrophy, hypertrophy, abnormal variation and fiber size), fiber splitting, vacuolar change, inflammatory infiltrates, and proliferation of connective tissue/fibrosis.39 A dystrophic myopathy frequently is characterized by the presence of normal fibers, hypertrophied fibers, degenerating fibers, atrophic fibers, regenerating fibers, and connective tissue and fatty infiltration (Figure 48-5).
Immunoblotting and Immunostaining
Immunoblotting of a muscle sample provides information about amounts of specific muscle protein, such as dystrophin or other structural proteins important in maintaining the structural integrity of the muscle membrane. Immunoblotting can be performed with as little as 10 mg of frozen tissue. Quantitative dystrophin analysis using Western blot technique can differentiate DMD from BMD and thus help determine the prognosis in a young symptomatic patient, which is not determined by standard molecular genetic analysis of the dystrophin gene. Dystrophin quantity 5% or less is consistent with DMD. Dystrophin levels of 5% to 20% of normal are seen in some with less severe “outlier” DMD or severe BMD, and either 20% to 80% dystrophin or normal quantity with reduced or increased molecular weight dystrophin is consistent with BMD. A normal dystrophin level in a patient with histologic evidence of a dystrophic myopathy is suggestive of LGMD. Immunofluorescent staining of muscle biopsy sections for dystrophin helps identify symptomatic female DMD carriers and some female BMD carriers.
The progressive loss of muscle fibers evident in muscular dystrophy is now thought to be caused by primary muscle sarcolemmal membrane abnormalities resulting from inherited structural abnormalities (abnormal molecular weight, deficiency, or absence) of dystrophin or dystrophin-associated transmembrane glycoproteins. Membrane instability leads to membrane injury from mechanical stresses, transient breaches of the membrane, and membrane leakage. Irreversible muscle cell death occurs ultimately, after multiple cycles of degeneration and regeneration. The muscle fiber is then replaced by connective tissue and fat, and this fibrotic replacement of the muscle can be exceedingly aggressive. This has given rise to the concept of diseases of the dystrophin–glycoprotein complex. Primary genetic abnormalities lead to abnormalities of intracellular dystrophin, transmembrane sarcoglycans, or transmembrane dystroglycans. Specific muscle cytoskeletal protein abnormalities lead to specific dystrophic disease phenotypes. An abnormality in the muscle protein merosin, located in the extracellular matrix,31 gives rise to one of the forms of CMD. Immunoblotting and immunofluorescent staining of the proteins of the dystrophin–glycoprotein complex now allow many LGMD patients to be subtyped.
Classification of Myopathies
Myopathies can be classified as hereditary or acquired, and the specific etiologies can be varied as shown in Box 48-2. The following descriptions pertain to some of the more common myopathies encountered in clinical practice.
Specific Hereditary Myopathies
Dystrophic Myopathies
“Muscular dystrophies” are debilitating myopathic disorders that present with muscle wasting and diffuse muscle weakness. They are caused by genetic mutations that produce muscle fiber necrosis and regeneration, ultimately resulting in muscle fiber loss. Patients with muscular dystrophies were traditionally grouped together because they had similar pathologies. They were subdivided into categories based on their modes of inheritance, ages of onset, and distributions of affected muscles. Most types of muscular dystrophy are not purely muscle disorders but rather multisystem disorders with disease manifestations in a variety of body systems, which can include the musculoskeletal, cardiovascular, pulmonary, and gastrointestinal systems, as well as endocrine system, skin, eyes, brain, and other organ systems. Muscular dystrophies are caused by mutations of the genes encoding for proteins important for the stability of the sarcolemmal membrane and the maintenance of muscle fiber intracellular homeostasis. They are diverse diseases genetically, biochemically, and clinically.
Dystrophinopathies
Duchenne Muscular Dystrophy
DMD is an X-linked disorder caused by an abnormality at the Xp21 gene loci. The gene for DMD and BMD occupies 2.5 million base pairs of DNA on the X chromosome and is about 10 times larger than the next largest gene identified to date. The gene coding sequence contains 79 exons. Its primary protein product, dystrophin, is localized to the intracellular side of the plasma membrane of all myogenic cells, certain types of neurons, and in small amounts of other cell types. Dystrophin deficiency at the plasma membrane of muscle fibers disrupts the membrane cytoskeleton and leads to the secondary loss of other components of the muscle cytoskeleton. The primary consequence of the cytoskeleton abnormalities is membrane instability, leading to membrane injury from mechanical stresses, transient breaches of the membrane, and membrane leakage. Chronic dystrophic myopathy is characterized by aggressive fibrotic replacement of the muscle and eventual failure of regeneration with muscle fiber death and fiber loss. Generally loss of the reading frame causes complete absence of dystrophin and a Duchenne phenotype. For cases with a deletion mutation, the “reading frame” hypothesis predicts that BMD patients with in-frame deletions produce a semifunctional, internally deleted dystrophin protein. DMD patients with frameshift or “out of frame deletions,” on the other hand, produce a severely truncated protein that is unstable.72 Characteristics of DMD and BMD are shown in Table 48-2.
Table 48-2 Characteristics of Dystrophinopathies (DMD and BMD)
| DMD | BMD | |
|---|---|---|
| U.S. prevalence (estimated) | 15,000 | 3700-8300 |
| Incidence rate | 1/3500 Male births | Unknown |
| Inheritance | X-linked | X-linked |
| Gene location | Xp21 (reading frame shifted) | Xp21 (reading frame maintained) |
| Protein | Dystrophin | Dystrophin |
| Onset | 2-6 yr | |
| Severity and course | ||
| Ambulation status | Loss of ambulation >16 yr | |
| Weakness | ||
| Cardiac | Cardiomyopathy (can occur before weakness); third to fourth decade frequent | |
| Respiratory | ||
| Muscle size | Calf hypertrophy | Calf hypertrophy |
| Musculoskeletal | Contractures: ankles and others in adulthood | |
| Central nervous system | Some patients have reduced cognitive ability | |
| Muscle pathology | ||
| Blood chemistry and hematology |
ALT, Alanine aminotransferase; AST, aspartate aminotransferase; BMD, Becker muscular dystrophy; CK, creatine kinase; DMD, Duchenne muscular dystrophy; GGT, γ-glutamyl transpeptidase.
Onset, Early Signs, and Disease Progression
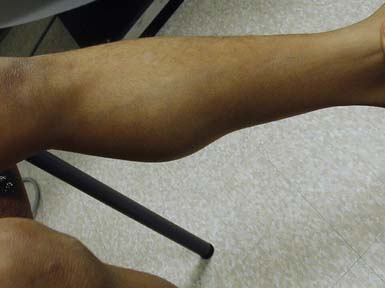
In most cases, the onset of DMD is noted before 4 to 5 years of age.84,85 The most frequent presenting signs and symptoms are abnormal gait with toe walking, lumbar lordosis, and proximal weakness. Difficulty negotiating stairs is an early feature as is a tendency to fall as a result of the child tripping or stumbling on a plantar-flexed ankle or the knee buckling or giving way because of knee extensor weakness. There is progressive difficulty getting up from the floor with presence of Gower’s sign. Parents frequently note the toe walking, which is a compensatory adaptation to knee extensor weakness. They also note a lordotic posture of the lumbar spine, which is a compensatory change resulting from hip extensor weakness. Pain in the muscles, especially the calves, is a common symptom. Enlargement of muscles as a result of pseudohypertrophy, particularly the calves (Figure 48-6), is commonly seen. The tongue is also frequently enlarged. The earliest weakness is seen in the neck flexors during preschool years. Early in the disease course, weakness is generalized but predominantly proximal. Pelvic girdle weakness predates shoulder girdle weakness by several years. Ankle dorsiflexors are weaker than ankle plantar flexors; ankle everters are weaker than ankle inverters; knee extensors are weaker than knee flexors; hip extensors are weaker than hip flexors; and hip abductors are weaker than hip adductors.85 The weakness progresses steadily, but the rate can be variable during the disease course. The average age of full-time wheelchair use in a DMD population not treated with corticosteroids has been age 10 with a range of 7 to 13. Timed motor performance is useful for the prediction of time when ambulation will be lost without provision of long-leg braces. One large natural history study showed that all DMD subjects who took 12 seconds or longer to ambulate 30 feet lost ambulation within 1 year.85 Ambulation past the age of 14 in a non–corticosteroid-treated patient should raise the suspicion of a milder form of muscular dystrophy such as BMD or LGMD. Ambulation beyond 16 years has been previously used as an exclusionary criterion for DMD in studies of BMD. Immobilization for any reason can lead to a marked and often precipitous decline in muscle power, rapid development of contractures, and loss of ambulatory ability. A fall with resultant fracture leading to immobilization and loss of ambulatory ability is not an uncommon occurrence.
Pharmacologic Treatment
Treatment with prednisone or deflazacort (not approved by the Food and Drug Administration in the United States) helps maintain strength and prolongs ambulation by 2 years.79,91 There does not appear to be an advantage of deflazacort over daily prednisone. The optimal dose of prednisone is 0.75 mg/kg/day up to a maximum of 40 mg/day.29,79,91 The optimal dose of deflazacort appears to be 0.90 mg/kg/day. With both corticosteroid regimens the patients need to be monitored for cataracts, hypertension, weight gain, osteoporosis, growth retardation, diabetes, and behavioral side effects.
Contractures
Significant joint contractures have been found in nearly all DMD children older than age 13.20,67,85 The most common contractures include ankle plantar flexion, knee flexion, hip flexion, iliotibial band tightness, and elbow flexion and wrist flexion contractures.85 Mild shortening of the iliotibial bands, hip flexor muscles, and heel cords occur in most DMD patients by 6 years of age.19 The presence of lower limb contractures in DMD has been shown to be strongly related to onset of wheelchair reliance.85 Lower extremity equinus contractures occur while DMD subjects are still upright, but knee and hip contractures are mild or absent in the ambulatory patient. Hip, knee, and elbow flexion contractures develop soon after DMD patients assume a sitting position in a wheelchair for most of the day as a result of prolonged static positioning of the limb with fibrotic musculature. Few subjects exhibit plantar flexion contractures of 15 degrees or more before their transition to a wheelchair.85 Natural history data suggest that weakness is the major cause of loss of ambulation in DMD, not contracture formation.
Spine Deformity
The reported occurrence of significant scoliosis in DMD subjects not treated with corticosteroids varies from 33% to 100%.51 This marked variability is primarily because of retrospective selection for scoliosis, the inclusion or exclusion of functional curves, and dissimilar age-groups. The prevalence of scoliosis is strongly related to age. Fifty percent of DMD patients acquire scoliosis between ages 12 and 15, corresponding to the adolescent growth spurt. In a non–corticosteroid-treated cohort, Oda et al.98 reported that 15% of older DMD patients show mild nonprogressive curves (usually 10 to 30 degrees). The likelihood of severe progressive spinal deformity could be predicted by type of curve and early pulmonary function measurements. Those with spines lacking significant kyphosis or hyperlordosis and a peak obtained absolute forced vital capacity (FVC) greater than 2000 mL tended not to show severe progressive scoliosis.98 No cause-and-effect relationship has been established between onset of wheelchair reliance and occurrence of scoliosis.76,85 Wheelchair reliance and scoliosis have both been found to be age-related phenomenon and likely represent coincidental signs of disease progression.
In retrospective series, treatment of DMD with deflazacort and prednisone has been shown to reduce the occurrence of significant scoliosis.7,11,60 It remains to be seen whether the apparent arrest in the development of scoliosis with corticosteroids will continue past the age of skeletal maturity. For curves that have progressed past 20 to 40 degrees, spinal arthrodesis has been shown to be the only effective treatment. Spinal orthoses are generally not used for preventing scoliosis in those with DMD.
Pulmonary Function
Absolute FVC volumes increase in DMD during the first decade and plateau during the early part of the second decade.85 A linear decline in percentage of predicted FVC is apparent between 10 and 20 years of age in those with DMD.85 Rideau et al.106 reported FVC to be predictive of the risk of rapid scoliosis progression. In the most severe DMD cases, maximal FVC reached a plateau of less than 1200 mL. This was associated with loss of ability to walk before age 10 and severe progressive scoliosis. McDonald et al.85 found that those patients with higher peak FVC (>2500 mL) had a milder disease progression, losing 4% predicted FVC per year. Those with peak predicted FVC less than 1700 mL lost 9.6% predicted FVC per year. Prednisone and deflazacort both appear to reduce the loss of pulmonary function over time during the second decade in DMD.7,11,30,60
Maximal static airway pressures (both maximal inspiratory pressure and maximal expiratory pressure) are the earliest indicators of restrictive pulmonary compromise in those with DMD, with impaired values noted between 5 and 10 years of age. DMD patients typically show a linear decline in percentage of predicted FVC during the second decade. An FVC less than 35% is associated with increased perioperative morbidity in DMD,89 and surgery ideally should be performed in those with percentage of predicted FVC greater than 40%. Recent evidence suggests that spinal arthrodesis can be safely performed in a population of DMD patients with percentage of predicted vital capacity less than 30%.80
Cardiomyopathy
The dystrophin protein is present in both the myocardium and the cardiac Purkinje fibers. Abnormalities of the heart can be detected by clinical examination, electrocardiogram (ECG), echocardiography, and Holter monitoring. Nearly all patients over the age of 13 demonstrate ECG abnormalities.85 Q waves in the lateral leads are the first abnormalities to appear, followed by elevated ST segments and poor R-wave progression, increased R/S ratio, and finally resting tachycardia and conduction defects. Localized posterior wall fibrosis was found to be peculiar to DMD and was not found in other types of muscular dystrophy.135 Sinus tachycardia can be due to low stroke volume from the progressive cardiomyopathy, or in some cases can be sudden in onset and labile, suggesting autonomic disturbance or direct involvement of the sinus node by the dystrophic process.88
Clinically evident cardiomyopathy is usually first noted after age 10 and is apparent in nearly all patients over age 18.96 Echocardiography has been used extensively to follow the development of cardiomyopathy and predict prognosis in patients with DMD. The onset of systolic dysfunction noted by echocardiography is associated with a poor short-term prognosis.96 The myocardial impairment remains clinically silent until late in the course of the disease, possibly caused by the absence of exertional dyspnea, secondary to lack of physical activity. Death has been attributed to congestive heart failure in as many as 40% to 50% of patients with DMD by some investigators.96 Ventricular ectopy and sudden death are known complications of the cardiomyopathy in DMD, and this association likely explains observed cases of sudden death.136 Regular cardiac evaluations with an ECG, echocardiography, and Holter monitor should be used in teenagers with preclinical cardiomyopathy.
Recent studies suggest that early presymptomatic treatment to achieve afterload reduction with angiotensin-converting enzyme (ACE) inhibitors such as perindopril or enalapril delayed the onset and progression of prominent left ventricle dysfunction in children with DMD.38 In another series, 43% with impaired left ventricular systolic dysfunction responded to enalapril with the normalization of function.104 Alternatively, angiotensin II type 1 receptor blockers (ARBs) such as losartan can be considered for afterload reduction in DMD. Animal studies show that the ARB losartan attenuates transforming growth factor–β-induced failure of muscle regeneration in dystrophinopathy, presenting an additional potential for therapeutic benefit vis-à-vis skeletal muscle in DMD.32
Cognition and Behavioral Phenotype
A dystrophin isoform is present in the brain. Previous studies on intellectual function in children with DMD have generally shown decreased intelligence quotient (IQ) scores when these children are compared with normative groups.85 A mean score for the DMD population of 1.0 to 1.5 standard deviations (SDs) below population norms has been reported. There has generally been consistency in the degree of impairment across measures reflecting a rather mild global deficit. Some studies37,55,57 have demonstrated relative deficits in verbal IQ and verbal memory. In a longitudinal assessment of cognitive function, McDonald et al.85 found IQ measure in those with DMD to be stable over time. On neuropsychologic testing, a large proportion of DMD subjects fell within the “mildly impaired” or “impaired” range according to normative data.85 Again, there were no particular areas of strength or weakness identified. These findings likely reflect a mild global deficit rather than focal nervous system impairment.85 An increased incidence of autism spectrum disorder has been found in those with DMD.134 In one large series of DMD subjects, 11.7% were reported to have a comorbid diagnosis of attention deficit hyperactivity disorder, 3.1% had autism spectrum disorder, and 4.8% had obsessive–compulsive disorder.54 In addition, impaired facial affect recognition has been found to be a part of the phenotype associated with DMD.56
Anthropometric Changes
Substantial anthropometric alterations have been described in those with DMD. Short stature and slow linear growth with onset shortly after birth have been reported.105 Arm span measurement, forearm segment, and radius length have been proposed as alternative linear measurements of growth in DMD patients with upper limb contractures. Obesity is a substantial problem in those with DMD, especially after the loss of independent ambulation.85,130 Weight control during early adolescence has its primary rationale in ease of care, especially in regard to ease of transfers during later adolescence. Immediately after spine fusion there has been a documented tendency for DMD patients to lose significant weight. The weight loss after surgery was associated with loss of self-feeding.62 No association with weight loss and loss of biceps strength was noted. A correction of the kyphosis might make self-feeding problematic in those with DMD. A feeding evaluation and incorporation of kyphosis into the spinal instrumentation construct can help preserve self-feeding and prevent weight loss subsequent to spine fusion.
Longitudinal weight measurements in older teenagers and young adults with DMD confirm significant rates of weight loss.85,130 This is likely caused by relative nutritional compromise during the later stages when boys with DMD have higher protein and energy intake requirements because of hypercatabolic protein metabolism. During the later stages of DMD, protein and calorie requirements can often be as high as 160% of that predicted for able-bodied populations.99,100 Restrictive lung disease becomes more problematic during this time, and this can also influence caloric intake and requirements. Self-feeding often becomes impossible during this period because of significant biceps weakness. Boys with DMD can develop signs and symptoms of upper gastrointestinal dysfunction in late adolescence.63
Becker Muscular Dystrophy
Existence of a form of muscular dystrophy with a similar pattern of muscle weakness seen in DMD, X-linked inheritance, but with later onset and a much slower rate of progression, was first described by Becker and Kiener in 1955.8 The disorder has the same gene location as the DMD gene (Xp21) and is allelic. On immunostaining of muscle biopsy specimens, the presence of a patchy abundance of dystrophin suggests a BMD phenotype. On Western blot for quantitative dystrophin analysis, either 20% to 80% dystrophin levels or normal quantity and reduced or increased molecular weight dystrophin is consistent with BMD. Studies show that 5% to 20% dystrophin quantity is consistent with an outlier or intermediate phenotype.58
Molecular Genetics and Diagnostic Evaluation
Full gene sequencing of the dystrophin gene demonstrates large deletions, duplications, and point mutations, identifies 98% to 99% of patients with dystrophinopathy, and is now the standard of care. This is essential for identification of specific gene alterations that will be targeted for molecular-based therapies. Differential diagnosis between DMD and BMD is best done by consideration of clinical findings, family history of clinical phenotype, and muscle biopsy with quantitative dystrophin analysis. Mutations at the Xp21 locus that maintain the translational reading frame (in-frame mutations) result in an abnormal but partially functional dystrophin protein. In DMD, however, the mutations shift the reading frame (out-of-frame mutations) so that virtually no dystrophin is produced. Absent dystrophin or levels less than 5% of normal generally are considered diagnostic of DMD,58 although 5% of such patients have BMD phenotypes. All BMD patients with normal molecular weight dystrophin have decreased quantities, usually less than 30% normal.
Age of Onset and Presenting Signs
Studies have shown significant overlap in the observed age of onset between DMD and BMD.84 A series by Bushby et al.23 that included 67 BMD subjects supported the presence of two major patterns of progression in BMD—a “typical” slowly progressive course and a more “severe” and rapidly progressive course. All the “severe” BMD cases showed difficulty climbing stairs by age 20, whereas none of the “typical” BMD cases had difficulty climbing stairs before age 20. Abnormal ECGs were seen in 27% of typical BMD subjects and 88% of severe subjects. Bushby et al.23 found BMD subjects to have a mean age of onset of 12 years in the typical group and 7.7 years in the severe group. Some adult patients with BMD present with major muscle cramps as an isolated symptom, and no weakness.22 As in DMD, preclinical cases are often identified by the finding of a grossly elevated CK value. A considerable overlap in CK values also exists between DMD and BMD cases at the time of presentation. Because of this, CK values cannot be used to differentiate DMD from BMD. Calf enlargement is a nonspecific finding in BMD, as is the presence of a Gower’s sign. The gait over time is similar to other neuromuscular disease conditions with proximal weakness. Patients often ambulate with a lumbar lordosis, forefoot floor contact, decreased stance-phase knee flexion, and a Trendelenburg or gluteus medius lurch (often described as a waddle).
Clinical Findings and Pattern of Progression
BMD patients have a distribution of weakness that is similar to those with DMD.84 The muscle groups that are most severely involved earlier in the course of disease include the hip extensors, knee extensors, and neck flexors.84 The most useful clinical criterion to distinguish BMD from DMD is the continued ability of the patient to walk into late teenage years. Those with BMD typically remain ambulatory beyond 16 years of age. Some might become wheelchair users in their late teens or 20s, whereas others can continue walking into their 40s, 50s, or later. DMD cases usually stop ambulating by 13 years unless treated with corticosteroids. Outlier DMD or intermediate dystrophinopathy cases generally stop ambulating between 13 and 16 years of age if untreated with corticosteroids. Early development of contractures does not appear to be a feature of BMD,22,23,84 but nonambulatory BMD subjects can develop equinus contractures, knee flexion contractures, and hip flexion contractures. Spinal deformity is not nearly as common or severe in BMD as it is in DMD. Spinal instrumentation is rarely required by DMD patients.22,23,84
Pulmonary Function
Compromised pulmonary function is much less problematic in patients with BMD than it is in those with DMD.22,84,106 The percentage of predicted FVC does not appear substantially reduced until the third to fourth decade. The percentage of predicted maximal expiratory pressure appears relatively more reduced at younger ages than the percentage of predicted maximal inspiratory pressure, a finding seen in those with DMD and other neuromuscular diseases.26,27,85,110
Cardiomyopathy
The pattern of occasional life-threatening cardiac involvement in otherwise mild and slowly progressive BMD has been reported by many.22,74 A significant percentage of BMD cases develop cardiac abnormalities, and the rate of progression of cardiac failure can on occasion be more rapid than the progression of skeletal myopathy.74 Approximately 75% of BMD patients have been found to have ECG abnormalities.84,114 The abnormal findings most typically reported include abnormal Q waves, right ventricular hypertrophy, left ventricular hypertrophy, right bundle branch block, and nonspecific T-wave abnormalities. On echocardiography, 63% have subnormal systolic function because of global hypokinesia.114 The cardiac compromise can be disproportionately severe compared with the degree of restrictive lung disease in some BMD subjects. The slowly progressive nature of this dystrophic myopathy, which is compatible with many years of functional mobility and longevity, makes these patients suitable candidates for cardiac transplantation if end-stage cardiac failure occurs.
Limb–Girdle Muscular Dystrophy
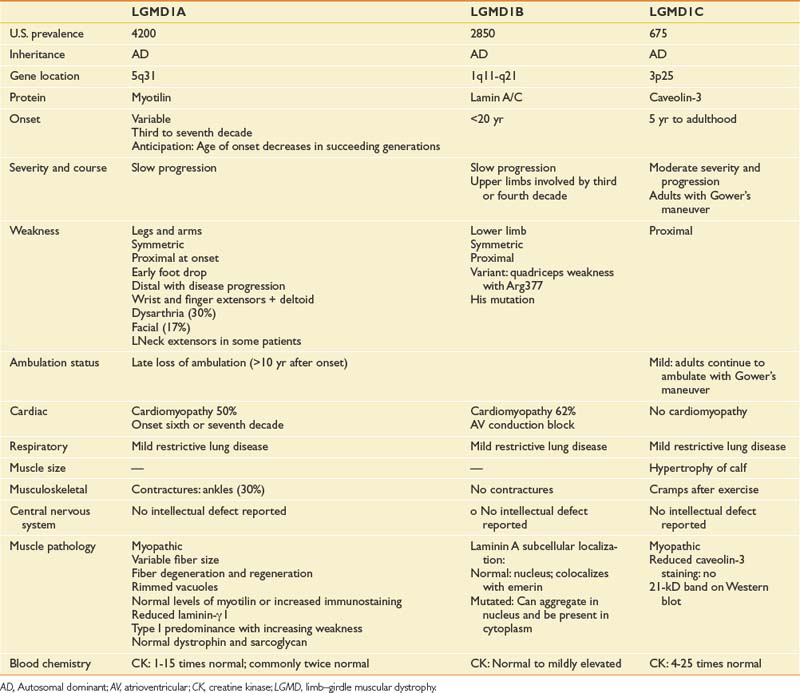
Before the advent of genetic testing, a group of patients commonly sharing a progressive pattern of greater proximal than distal muscular weakness with either autosomal dominant (LGMD1) or autosomal recessive (LGMD2) inheritance were said to have limb–girdle muscular dystrophies. Recent advances in molecular and genetic analyses have now identified a number of distinct genetic mutations in these patients. Those with LGMD1 subtypes usually have later onset in adulthood, whereas those with LGMD2 subtypes usually have onset during childhood or adolescence, although some can present in early adulthood. Many of the LGMD2 subtypes have been linked to gene defects causing abnormalities of the sarcolemmal-associated proteins, including sarcoglycans (α-SG, γ-SG, β-SG, and δ-SG), dystroglycans, calpain-3, dysferlin, fukutin-related protein (FKRP), telethonin, and titin. The most common LGMD2 subtypes include sarcoglycanopathies, dysferlinopathies, calpainopathies, and FKRP deficiencies. The distribution and pattern of weakness at onset are predominantly in the pelvic or shoulder girdle musculature or both. The rate of progression is slower in those with LGMD than in those with DMD.10,87,97 Clinical features of autosomal dominant LGMD1 subtypes are shown in Table 48-3. Clinical features of the most common forms of autosomal recessive LGMD2 are shown in Table 48-4.
< div class='tao-gold-member'>






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


