The relative contribution and source of the fuels used during endurance exercise is dependent on the intensity and the duration of the exercise. Much work has been done to investigate the potential performance-enhancing effect of manipulating training and dietary interventions in athletes, as well as the influence of gender. Studies show that even patients with metabolic myopathies may derive benefits that counter the age-associated loss of muscle mass and strength. This article gives an overview of these different impacts on endurance exercise, concluding with an examination of the metabolic myopathies that impair substrate metabolism in skeletal muscle and result in exercise intolerance.
Carbohydrate (CHO) and fat constitute the main fuels used during endurance exercise, while amino acid oxidation (protein) contributes a small amount to total substrate use. The relative contribution and source of the fuels used during endurance exercise is dependent on the intensity and the duration of the exercise bout. Additionally, the relative contribution of CHO, fat, and amino acids to fuel endurance exercise can be influenced by training status, sex, and diet. As such, much work has been done to investigate the potential performance enhancing effect of manipulating training and dietary interventions in athletes.
Given that acute exercise represents a metabolic challenge to intermediary metabolism, it is often a trigger of symptoms for individuals with disorders of carbohydrate and fat oxidation. Because the mitochondria are the final common pathway for oxidative metabolism of all metabolic substrates, patients with primary mitochondrial cytopathies also display symptoms of exercise intolerance. Pre-exercise carbohydrate ingestion is of therapeutic benefit for people with McArdle’s disease and fat oxidation defects, while carbohydrate loading strategies are helpful in the symptomatic treatment of fat oxidation defects. Conversely, triglyceride infusion can improve exercise capacity in patients with a complex I deficiency; yet a higher fat diet does not appear to benefit patients with mitochondrial complex I deficiency. Endurance exercise training is of significant benefit to those with McArdle’s disease and mitochondrial cytopathies. Resistance exercise training may also be of benefit for people with sporadic mitochondrial cytopathies. Finally, with the benefits of resistance exercise training being well documented in older adults, all patients with metabolic myopathies may derive benefits that counter the age-associated loss of muscle mass and strength.
Carbohydrate oxidation during endurance exercise
Carbohydrates are one of the main sources of fuel used during moderate to higher intensity endurance exercise. CHO are a versatile fuel, contributing at both aerobic and anaerobic power outputs. The CHO stores used during endurance exercise include plasma glucose and muscle glycogen. Muscle glycogen stores represent the major source of carbohydrates used during exercise under most circumstances, and are depleted by 1 to 2 hours of moderate to higher intensity exercise. Plasma glucose is derived endogenously from liver glycogen stores or from gluconeogensis in the liver and kidneys, with exogenous sources possible from dietary ingestion. The relative contribution of plasma glucose and muscle glycogen is influenced by exercise duration and intensity, training status, sex, and diet (see below). The uptake of plasma glucose into skeletal muscle is mediated by the facilitated transporters, GLUT-1 and GLUT-4. GLUT-1 is constitutively expressed at the sarcolemma, while GLUT-4 is translocated to the sarcolemma and transverse tubules via stimulation by insulin or exercise. Once glucose has entered the muscle it is phosphorylated by hexokinase II to glucose-6-phosphate (G6P), and can either undergo glycogen synthesis or enter glycolysis ( Fig. 1 ).
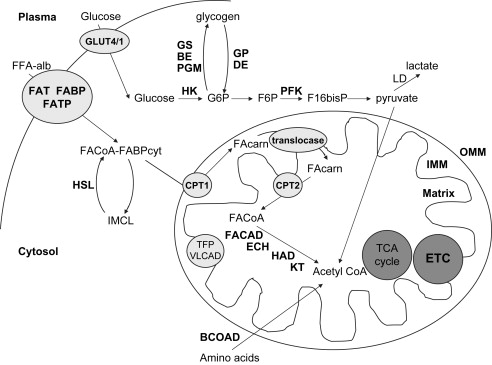
Glycogen synthesis begins with the conversion of G6P to glucose-1-phosphate by phosphoglucomutase, which is then converted into UDP-glucose by UDP-glucose pyrophosphorylase. UDP-glucose donates a glucosyl group, which glycogen synthase (GS, the rate-limiting enzyme in glycogen synthesis) and branching enzymes add to the glycogen granule via α-1,4- and α-1,6-glycosidic linkages, respectively. At the core of the glycogen granule is a protein primer, glycogenin, which acts as a primer for GS to form new glycogen granules. Skeletal muscle glycogen is stored in two main forms called pro- and macroglycogen, with proglycogen representing smaller units of glycogen with a relatively high associated protein content, while macroglycogen represents larger particles with a relatively high carbohydrate-to-protein content. Glycogen degradation begins with the conversion of glycogen to glucose-1-phosphate, catalyzed by glycogen phosphorylase and debranching enzyme, which cleave the α-1,4- and α-1,6-glycosidic linkages, respectively.
During endurance exercise, plasma glucose uptake increases because of increased translocation of GLUT-4 to the sarcolemma and T-tubules. The mechanisms of exercise-stimulated GLUT-4 translocation are unknown; however, they are different from insulin stimulated GLUT-4 translocation, and although still controversial, it is likely that AMP-activated protein kinase and calcium are involved. During exercise, most of the glucose that enters the muscle is routed toward glycolysis, which results in the production of pyruvate. Glycolysis is primarily regulated by phosphofructokinase (PFK), which is allosterically modified by activators such as inorganic phosphate and AMP, and inhibited by high levels of adenosine 5′-triphosphate. During exercise, pyruvate is either shunted into the tricarboxylic acid cycle by pyruvate dehydrogenase (PDH) under aerobic conditions, or toward lactate formation by lactate dehydrogenase when the rate of glycolysis exceeds that of PDH entry into the tricarboxylic acid cycle (such as at the onset of exercise or under ischemic conditions). The PDH complex is activated by a phosphatase and inhibited by a kinase (PDK). Dichloroacetate is an activator of PDH and has been used in some cases of mitochondrial cytopathy to reduce lactate levels.
Lipid oxidation during endurance exercise
Lipid oxidation contributes maximally during moderate-intensity exercise (40%–65% maximal oxygen consumption per unit time or VO 2 max; see the section on exercise intensity during endurance exercise, below), representing between 40% and 60% of total energy expenditure. Of this, plasma free fatty acids (FFA) contribute between 40% and 60% of total fat oxidation; other fat sources, intramyocellular lipids (IMCL, Fig. 2 ) and lipoprotein-derived triacylglycerol (TG), provide the remainder.

Under normal dietary conditions, the contribution of lipoprotein-derived TG during exercise is less than 10%, implying that IMCL supplies the remaining 30% to 50% of fat oxidation during moderate intensity endurance exercise. However, the use of IMCL during endurance exercise is controversial. Numerous studies have found that IMCL is an important substrate during moderate-intensity endurance exercise, whereas other studies have not found this. Additionally, several studies have found that only women use IMCL during endurance exercise. However, although it is plausible for women to use more IMCL during a bout of endurance exercise (see section below describing the effect of sex on substrate selection during endurance exercises), it is unlikely that men do not use any IMCL as a fuel source during endurance exercise.
The most likely cause of the aforementioned discrepancy relates to methodologic limitations associated with quantifying IMCL content. There are several methods used to quantify IMCL content, including biochemical extraction, stable isotope infusion, light microscopy, electron microscopy, and proton magnetic resonance spectroscopy ( 1 H-MRS). It is generally accepted that when using the biochemical method, there can be significant contamination from extramyocellular lipids. The majority of studies that have not shown a decrease in IMCL following a bout of endurance exercise have used the biochemical method, whereas studies using stable isotope tracers, electron microscopy (EM), and 1 H-MRS, consistently show a decrease in IMCL following a bout of endurance exercise for men and women. Furthermore, MRS and EM appear to show similar findings when directly compared. The use of more sophisticated methodologies ( 1 H-MRS and EM) has decreased the confusion regarding the use of IMCL during endurance exercise and it is now generally accepted that IMCL are an important fuel source during endurance exercise.
Free fatty acids are chaperoned in the plasma by albumin. There are three fatty acid binding proteins (fatty acid binding protein of the plasma membrane or FABPpm, fatty acid translocase or FAT/CD36, and fatty acid transporter or FAT) that are responsible for the uptake of FFA into the muscle. Once in the muscle, FFA are chaperoned by FABPcytosol (once charged with a CoA group, FACoA) and can be esterified into IMCL for storage and hydrolyzed when needed. During exercise there is an increase in plasma FFA uptake by the muscle as well as an increased IMCL hydrolysis mediated by hormone sensitive lipase (HSL), which is stimulated by epinephrine.
It is important to note that although there is a net decrease in IMCL during a bout of endurance exercise, IMCL hydrolysis and FFA esterification are in a state of continual flux during exercise. To be oxidized, FACoA must enter the mitochondria where they undergo β-oxidation (see Fig. 1 ). While short and medium chain FACoAs can diffuse into the mitochondria, long chain FACoAs (LCFACoA) require modification by carnitine palmitoyl transferase I and II (CPT I and CPT II) to enter the mitochondrial matrix. The activity of CPT I is inhibited by malonyl CoA, which is involved in FA synthesis. New research also suggests that FAT/CD36 might also play a role in LCFACoA transport into the mitochondrial matrix. Once in the mitochondrial matrix, the FACoA can undergo β-oxidation, which is a sequence of steps resulting in the formation of acetyl CoA and a FACoA with two less carbons (see Fig. 1 ). The acetyl CoA formed during each turn of β-oxidation then enters the citric acid cycle and the FACoA returns to the start of β-oxidation. Very long chain FAs undergo a special form of β-oxidation using very long chain acyl CoA dehydrogenase (VLCAD) and trifunctional protein at the inner mitochondrial membrane (see Fig. 1 ). VLCAD initiates β-oxidation and trifunctional protein combines the functions of the remaining three enzymes of β-oxidation with long chain specificity.
Lipid oxidation during endurance exercise
Lipid oxidation contributes maximally during moderate-intensity exercise (40%–65% maximal oxygen consumption per unit time or VO 2 max; see the section on exercise intensity during endurance exercise, below), representing between 40% and 60% of total energy expenditure. Of this, plasma free fatty acids (FFA) contribute between 40% and 60% of total fat oxidation; other fat sources, intramyocellular lipids (IMCL, Fig. 2 ) and lipoprotein-derived triacylglycerol (TG), provide the remainder.

Under normal dietary conditions, the contribution of lipoprotein-derived TG during exercise is less than 10%, implying that IMCL supplies the remaining 30% to 50% of fat oxidation during moderate intensity endurance exercise. However, the use of IMCL during endurance exercise is controversial. Numerous studies have found that IMCL is an important substrate during moderate-intensity endurance exercise, whereas other studies have not found this. Additionally, several studies have found that only women use IMCL during endurance exercise. However, although it is plausible for women to use more IMCL during a bout of endurance exercise (see section below describing the effect of sex on substrate selection during endurance exercises), it is unlikely that men do not use any IMCL as a fuel source during endurance exercise.
The most likely cause of the aforementioned discrepancy relates to methodologic limitations associated with quantifying IMCL content. There are several methods used to quantify IMCL content, including biochemical extraction, stable isotope infusion, light microscopy, electron microscopy, and proton magnetic resonance spectroscopy ( 1 H-MRS). It is generally accepted that when using the biochemical method, there can be significant contamination from extramyocellular lipids. The majority of studies that have not shown a decrease in IMCL following a bout of endurance exercise have used the biochemical method, whereas studies using stable isotope tracers, electron microscopy (EM), and 1 H-MRS, consistently show a decrease in IMCL following a bout of endurance exercise for men and women. Furthermore, MRS and EM appear to show similar findings when directly compared. The use of more sophisticated methodologies ( 1 H-MRS and EM) has decreased the confusion regarding the use of IMCL during endurance exercise and it is now generally accepted that IMCL are an important fuel source during endurance exercise.
Free fatty acids are chaperoned in the plasma by albumin. There are three fatty acid binding proteins (fatty acid binding protein of the plasma membrane or FABPpm, fatty acid translocase or FAT/CD36, and fatty acid transporter or FAT) that are responsible for the uptake of FFA into the muscle. Once in the muscle, FFA are chaperoned by FABPcytosol (once charged with a CoA group, FACoA) and can be esterified into IMCL for storage and hydrolyzed when needed. During exercise there is an increase in plasma FFA uptake by the muscle as well as an increased IMCL hydrolysis mediated by hormone sensitive lipase (HSL), which is stimulated by epinephrine.
It is important to note that although there is a net decrease in IMCL during a bout of endurance exercise, IMCL hydrolysis and FFA esterification are in a state of continual flux during exercise. To be oxidized, FACoA must enter the mitochondria where they undergo β-oxidation (see Fig. 1 ). While short and medium chain FACoAs can diffuse into the mitochondria, long chain FACoAs (LCFACoA) require modification by carnitine palmitoyl transferase I and II (CPT I and CPT II) to enter the mitochondrial matrix. The activity of CPT I is inhibited by malonyl CoA, which is involved in FA synthesis. New research also suggests that FAT/CD36 might also play a role in LCFACoA transport into the mitochondrial matrix. Once in the mitochondrial matrix, the FACoA can undergo β-oxidation, which is a sequence of steps resulting in the formation of acetyl CoA and a FACoA with two less carbons (see Fig. 1 ). The acetyl CoA formed during each turn of β-oxidation then enters the citric acid cycle and the FACoA returns to the start of β-oxidation. Very long chain FAs undergo a special form of β-oxidation using very long chain acyl CoA dehydrogenase (VLCAD) and trifunctional protein at the inner mitochondrial membrane (see Fig. 1 ). VLCAD initiates β-oxidation and trifunctional protein combines the functions of the remaining three enzymes of β-oxidation with long chain specificity.
Amino acid oxidation during endurance exercise
During endurance exercise there is an increase in leucine oxidation, contributing approximately 2% to 3% of total substrate oxidation during the exercise bout. Leucine is oxidized by branched-chain 2-oxoacid dehydrogenase (BCOAD) within the mitochondria (see Fig. 1 ). BCOAD is the rate-limiting enzyme in branched-chain amino acid (leucine, isoleucine, valine) oxidation and is activated upon dephosphorylation by a specific phosphatase and inactivated by BCOAD kinase. During endurance exercise, the activity of BCOAD increases because of increased dephosphorylation of the enzyme, and returns to baseline within 10 minutes after cessation of exercise. The mechanism by which BCOAD activity increases with exercise is unclear. It has been proposed that with muscle stimulation there is an increase in the concentration of α-ketoisocaproic acid, which inhibits BCOAD kinase by interrupting the protein-protein interaction between itself and BCOAD, allowing increased BCOAD activity. In addition to the branched chain amino acids, alanine, asparagine, aspartate, glutamate, and lysine also appear to be oxidized in skeletal muscle. The magnitude of increase in the oxidation for lysine during endurance exercise is not as great as that seen for leucine.
Exercise intensity and duration impact substrate selection during endurance exercise
Altering exercise intensity has a profound effect on substrate selection during endurance exercise. At low exercise intensities (up to approximately 48% VO 2 max), fat is the primary fuel used to sustain exercise. In a study conducted by Romijn and colleagues during exercise at 25% VO 2 max, fat oxidation accounted for 86% of total substrate oxidation. Similarly, when exercise was performed at approximately 40% VO 2 max, fat oxidation contributed 55% to total substrate oxidation, while CHO oxidation contributed 45%. As exercise intensity increases from mild to moderate, the rate of fat oxidation also increases, reaching a maximum between 48% VO 2 max and 65% VO 2 max, after which the rate of fat oxidation declines.
Venables and colleagues found that the maximal rate of fat oxidation occurred at 48% VO 2 max, and this roughly coincided with the “crossover point” of substrate use, where CHO becomes the predominant fuel used to support endurance exercise. Whereas fat oxidation reaches a maximum and then declines at higher exercise intensities, CHO oxidation continues to increase, with increasing exercise intensities contributing approximately 70% to total substrate oxidation at 85% VO 2 max.
Exercise intensity not only alters the relative contribution of fat and CHO to total substrate oxidation, it also influences the source of fat and CHO that are used during endurance exercise. At low exercise intensities where fat oxidation predominates, plasma fuel sources (FFA and glucose) are the main contributors to fuel use, while muscle stores (IMCL and glycogen) are not significant contributors. As exercise intensity increases, CHO sources contribute more to substrate oxidation, there is an increased reliance on muscle substrate stores, while the contribution of plasma stores remains fairly constant.
The mechanism behind the shift in fuel use with increasing exercise intensity remains unclear. However there are several putative mechanisms that may explain these changes, including changes in adipose tissue blood flow, cell signaling molecule phophorylation, muscle pH, and interactions between CHO and fat metabolism. The effect of exercise duration on substrate selection during endurance exercise is more straightforward. During low intensity exercise (25% VO 2 max) the relative contribution of fat and CHO remains constant, with plasma FFA being the main source of fuel used to fuel a 120 minute exercise bout. During moderate intensity exercise (65% VO 2 max), there is a shift to a greater fat oxidation as exercise duration increases because of a reduction in muscle glycogen stores as exercise progresses. Additionally, as exercise duration increases there is an increased reliance on plasma FFA and glucose and a decreased reliance on IMCL and muscle glycogen.
Effect of endurance training on substrate selection during endurance exercise
Endurance exercise training increases the reliance on lipid oxidation during exercise at any given absolute workload. Evidence as to the source of the increased reliance on lipid during endurance exercise following training is controversial. Several studies have found that training increases the reliance on plasma FFA. Alternatively, several studies have shown that endurance training results in an increased oxidation of nonplasma FFA sources (presumably IMCL, ) or greater IMCL depletion. Additionally, several studies have found that endurance training decreases plasma FFA Rd, and thus plasma FFA oxidation. Likewise, endurance training did not change adipose tissue lipolytic response to epinephrine infusion and also did not increase whole-body lipolytic response when exercise was performed at the same absolute intensity.
A potential reason for the increased reliance on plasma FFA following training seen in the aforementioned study could be the timing of testing. While this study tested subjects in the postabsorptive state, most studies finding an increased reliance on IMCL following training tested subjects following an overnight fast. However, similar to studies finding that training increased the reliance on IMCL following training, one study finding increased plasma FFA oxidation after training was also performed following an overnight fast. Further work is needed in this area to elucidate whether endurance training increases plasma FFA or IMCL oxidation or both.
Endurance training increases IMCL content and this is thought to be one of the early responses to endurance training. A recent study using electron microscopy identified that training-induced increases in IMCL content were the result of an increase in the number of IMCL droplets in a given area, not an increase in IMCL size. Metabolically, the effect of endurance training to increase IMCL number and not IMCL size is logical, as this would increase the surface area of IMCL available to interact with mitochondria. An additional finding of the aforementioned study was that endurance training increased the percent of IMCL in direct contact with mitochondria. These findings support the theory that there is an increased reliance on IMCL during endurance exercise following training.
An increased reliance on lipid stores following training is supported by the finding that endurance training increases the capacity of numerous enzymes important in lipid oxidation and mitochondrial function. Endurance training in humans increased the activity of enzymes involved in β-oxidation, including β-hydroxyacyl-CoA dehydrogenase and CPT I, as well as the protein content of several enzymes involved in β-oxidation, including VLCAD and medium chain acyl-CoA dehydrogenase (MCAD). Endurance training also enhanced FFA uptake capacity by the muscle, as evidenced by an increased FABPpm and FAT/CD36 protein content in rats. Additionally, endurance training in humans increased mitochondrial function by increasing mitochondrial content, citrate synthase, malate dehydrogenase, succinate cytochrome c oxidoreductase (CII and CIII), cytochrome c oxidase (COX), dehydrogenase (CII), and nicotinamide adenine dinucleotide H cytochrome c oxidoreductase (CI and CIII) enzyme activities. Together, these findings provide the molecular basis for the increased reliance on lipid stores in trained individuals when subjects are tested at the same absolute exercise intensity.
Endurance training also increases muscle glycogen content at rest. This elevated resting muscle glycogen content is caused by an increased rate of glycogen accumulation following a bout of endurance exercise when carbohydrate is provided. The rate of accumulation of glycogen is two times greater in trained, as compared with untrained individuals, during the first 6 hours following a bout of glycogen-depleting exercise. Additionally, 48 and 72 hours after the exercise bout, glycogen levels are 66% higher in trained individuals. The molecular mechanism behind the increased rate of glycogen accumulation with endurance training is thought to be mediated via increased GLUT-4 protein content. A cross-sectional study has shown that GLUT-4 protein content is three times higher in trained versus untrained individuals. In humans and rats, endurance training increased GLUT-4 protein content and insulin stimulated glucose uptake, with no effect on insulin receptor function. Specifically, with endurance training in rats, there is an increased number of glucose transporters and GLUT-4 protein content at the plasma membrane, not the microsomal membrane. Whether this distributional change is seen in humans is yet to be determined. The increase in GLUT-4 protein content is a very early response to endurance training, occurring within 7 days of training onset. The effect of detraining on GLUT-4 protein content and insulin stimulated glucose uptake occurs even more rapidly, being reversed within a few days of inactivity.
Endurance training not only increases glucose transport into the muscle at rest, but also glucose phosphorylation, as evidenced by an increased hexokinase II activity with endurance training. Following glucose phosphorylation, G6P can be routed toward glycolysis or storage as glycogen. The impact of endurance training on GS activity is at odds with several studies finding no effect of training on GS activity. Thus, it appears that endurance training increases glycogen accumulation by its effect on GLUT-4 and perhaps hexokinase II activity.
As there is an increased reliance on lipid sources following training, there is a reciprocal effect of endurance training on CHO use during exercise. Numerous studies have found that during exercise at the same absolute intensity there is a decreased reliance on CHO following a period of endurance training. Despite an increased GLUT-4 protein content and an increased insulin stimulated glucose uptake at rest following a period of endurance training, glucose uptake during a bout of endurance exercise is lower. Specifically, training results in a lower glucose Ra, Rd, and metabolic clearance rate (MCR), as well as decreasing net glucose uptake across the working limb during moderate intensity exercise. As a result of this lower glucose uptake, there is a lower oxidation of plasma glucose in the trained state, as evidenced by a reduced rate of plasma glucose oxidation. The effects of training on glucose Ra, Rd, and MCR are seen as soon as 10 days of training and continue to become more pronounced with increased training duration, up to 12 weeks of training. The decreased glucose Ra during exercise in the trained state is mainly the result of a decreased rate of hepatic glycogenolysis, but there is also an effect of endurance training on decreasing the rate of hepatic gluconeogenesis as well. Mechanistically, the decreased glucose uptake during exercise in the trained state is mainly the result of decreased GLUT-4 translocation to the sarcolemma, and thus a lower glucose uptake capacity.
Endurance training attenuates the use of muscle glycogen during a bout of moderate intensity exercise. However, during high intensity exercise the use of muscle glycogen is the same between trained and untrained individuals. This finding suggests that, although CHO is spared during exercise performed in the trained state, when exercise intensities increase and lipid is no longer the preferred fuel to support exercise, muscle glycogen begins to compensate for the increased metabolic demand. Support for this notion comes from the finding that plasma glucose uptake remains blunted in trained individuals during high intensity exercise, and that plasma glucose contributes less to total CHO oxidation following a period of endurance training. Endurance exercise training also reduces the proportionate oxidation of leucine during acute endurance exercise for both men and women. This latter effect is associated with a marked attenuation of the exercise-mediated activation of the BCOAD enzyme activity, in spite of an increase in total BCOAD enzyme content. Recently, it has been shown that the training induced attenuation of BCOAD activation and leucine oxidation is associated with an increase in the BCOAD kinase protein content, implying enzyme inactivation. The reduction in BCOAD activation is associated with higher total muscle glycogen levels and lower glycogenolysis after training.
Effect of sex on substrate selection during endurance exercise
Numerous studies have found that women have a lower respiratory exchange ratio (RER) during endurance exercise as compared with men. More specifically, women, as compared with men, use less muscle glycogen, have higher glycerol turnover, higher plasma FFA concentration, lower glucose Rd, lower leucine oxidation, and higher IMCL oxidation during endurance exercise. It has been suggested that these sex differences during exercise are because of differences in estrogen concentration and activity. In fact, estrogen supplementation studies in animals and humans have supported the aforementioned findings of higher fat oxidation for women during endurance exercise.
One of the major limiting factors affecting sex comparative studies comes from the ability to accurately match women and men. Men typically have lower percent body fat and a higher maximum oxygen consumption (VO 2 max), thus subject selection based on body composition and VO 2 max is not advisable. As such, it is suggested that men and women be selected based on training status, training quantity, and VO 2 per kilogram fat free mass, as these are better determinants of fuel use. Additionally, as menstrual cycle phase can impact fuel selection during exercise, it is important to take this into consideration when testing eumenorrheic women. Habitual dietary intake can also influence substrate use, with high fat diets increasing fat oxidation and high CHO diets increasing CHO oxidation during exercise. As such, sex differences in nutrient consumption can influence fuel use during exercise, impacting the ability to detect an effect of sex on fuel use during exercise. However, dietary differences do not likely impact the aforementioned sex differences, for several studies that have looked at habitual dietary intake in men and women have not found any differences in the relative contribution of CHO, fat, and protein to total energy intake.
A higher abundance of type I muscle fibers in women is likely a significant contributor to the observed sex differences in exercise metabolism. A sex difference in metabolism during endurance exercise was first suggested when women began to out perform men during ultra-endurance events. During these events women can sustain a higher %VO 2 max for a longer period of time, as compared with equally trained men. Since then, numerous studies have shown that women rely on a greater proportion of lipid to fuel endurance exercise, thus sparing carbohydrate stores, which allows women to maintain a higher %VO 2 max. The sources of the increased reliance on lipid and decreased reliance on CHO by women during exercise remains controversial. It is generally accepted that women have a lower glucose Ra, Rd, and MCR during moderate intensity endurance exercise as compared with men, implying a decreased rate of liver glycogenolysis/gluconeogenesis during exercise in women. However, the effect of sex on muscle glycogen use during exercise yields conflicting results. To date only two studies have found that women have a lower net use of muscle glycogen as compared with men, while others have found no effect of sex on muscle glycogen use. Differences in the exercise protocol used (running versus cycling) and the menstrual cycle phase the women were tested in may have contributed to the finding of decreased muscle glycogen use in the aforementioned studies.
The source of the increased reliance on lipid by women during exercise remains an area of controversy. A lower RER, thus a greater reliance on lipid stores during exercise, in women is confirmed by studies finding that women have a greater glycerol Ra and Rd, a marker of whole body lipolysis, as compared with men. However, whether the increased reliance on lipid stores by women during exercise is plasma- or muscle-derived is a matter of debate.
The effect of sex on IMCL use during exercise is also controversial. It has been consistently shown that women have a greater IMCL content as compared with men. Additionally, using the EM technique the authors’ laboratory has shown that before and after a period of endurance training women have a higher number of IMCL in a given area of muscle, but not a greater lipid size, as compared with men, and this greater number of IMCL contributed to the increased IMCL content in women. Thus, women have an increased availability of IMCL to use as a substrate source during exercise; however, whether this indeed occurs is a matter of controversy. Because the use of IMCL during endurance exercise is controversial, sex differences in IMCL use during exercise are hard to determine. Several studies have found that women use more IMCL during a bout of moderate intensity endurance exercise as compared with men; however, these later studies also found that men do not use any IMCL during the exercise bout. The results of the aforementioned studies may be influenced by the use of the biochemical method to determine IMCL content and net use. A study using 1 H-MRS found that men used more IMCL than women during exercise, but these findings are confounded by the fact that the men were more trained than the women, which would result in increased IMCL use during exercise. Other studies using 1 H-MRS and EM and using equally trained men and women have found no sex difference in IMCL use during exercise or no difference in IMCL use between the sexes. Collectively, these findings suggest that when men and women are equally trained there is no difference in net IMCL use during a bout of endurance exercise at 65% VO 2 max.
If the increased reliance on lipids by women during exercise is not a result of increased IMCL use, it is reasonable to assume that it is plasma derived. Yet, several studies have found no effect of sex on plasma FFA Ra uptake and oxidation during exercise performed at 25%, 45%, 65%, and 85% VO 2 max. However, one study did find that during the last 30 minutes of a 90 minute cycling bout at 50% VO 2 max, women oxidized more plasma FFA as compared with men. Additionally, although not significant, another study found that during the last hour of a 90 minute exercise bout at 60% VO 2 max, women oxidized 32% more plasma FFA as compared with men. The results of these two studies are complicated by the fact that during that time point there was no difference in RER between the sexes. Thus, the impact of sex on plasma FFA oxidation during exercise remains controversial.
Perhaps sex differences in lipoprotein-derived TG use exist. To date no study has investigated the effect of sex on lipoprotein-derived TG use during a bout of endurance exercise. However, a recent study showed that at rest in both the fasted and the fed state, women had a greater uptake of serum TG across the leg, as compared with men. Whether or not this would result in greater lipoprotein-derived TG use during a bout of endurance exercise in women is yet to be determined. Additionally, because lipoprotein-derived TG contribute only approximately 10% to total fat oxidation during exercise, the physiologic importance of this finding needs to be determined.
Few studies have been conducted investigating the molecular mechanism behind the observed sex differences in metabolism during exercise. From a mechanistic standpoint, the increased reliance on lipids by women during endurance exercise appears to be an increased capacity for women to uptake plasma FFA into the muscle, as evidenced by the finding of increased mRNA expression of FAT/CD36 and FABPpm (untrained only), as well as an increased FAT/CD36 protein content (untrained and trained) in muscle from women as compared with men. However, no sex difference in plasma FFA uptake during exercise has been observed.
Women also rely to a lesser extent on amino acid oxidation during endurance exercise as compared with men. Specifically, men have a higher rate of protein oxidation at rest and during exercise, and a higher leucine flux, while women have a higher rate of nonoxidative leucine metabolism. Combining data from six studies investigating the relative contribution of protein to total substrate oxidation during exercise, the authors’ laboratory has found that protein contributes 2.1% of total energy during exercise in women, while in men it contributes 5.5% ( P = .02).
The observed sex difference in metabolism is mediated by differences in estrogen concentration between the sexes. Supplementation trials of 17-β-estradiol (E2) in animals and humans have shown that short-term administration of E2 can modify fuel selection during endurance exercise. In humans, short term E2 supplementation decreased glucose Ra, Rd, MCR, and RER, with no effect of glycerol Ra and Rd. E2 supplementation also lowered resting muscle glycogen stores with no effect on muscle glycogen use. However, there does not appear to be an effect of E2 supplementation on performance during exercise lasting 1 to 2 hours. Collectively these data suggest that E2 has a primary action on liver glycogenolysis and glucose release, while muscle glycogenolysis is not affected by short term E2 supplementation. Additionally, these studies suggest that at the level of substrate use E2 has a greater impact on CHO, as opposed to lipid, metabolism. To date only one E2 supplementation study has been conducted investigating the effects of E2 on amino acid metabolism during endurance exercise. In this study, 8 days of high dose E2 supplementation (1 mg per day for 2 days; 2 mg per day for 6 days) to men lowered leucine oxidation at rest and during exercise. Additionally, E2 supplementation resulted in a less negative protein balance. However, there was no effect of E2 supplementation on leucine flux, protein synthesis, or protein breakdown.
Fluctuations in ovarian hormones across the menstrual cycle can also influence substrate use during exercise; however, this has not been corroborated in all studies. Women in the luteal phase of the menstrual cycle have a lower RER, CHO oxidation, glucose Ra, Rd, and muscle glycogen use, as well as higher lipid oxidation. However, not all studies have demonstrated a lower glucose Ra and Rd during the luteal phase of the menstrual cycle, a finding that may be explained by differences in the exercise intensity used or whether subjects were tested in the fed or fasted state. Additionally, it has been consistently shown that women in the luteal phase of the menstrual cycle use less muscle glycogen. The effect of the menstrual cycle on lipid metabolism is unclear. Several studies have found a lower RER as well as higher lipid oxidation during exercise performed by women in the luteal phase of the menstrual cycle. However, to date no effect of menstrual cycle phase has been found on glycerol or palmitate flux. To the best of the authors’ knowledge, no study to date has investigated the impact of menstrual cycle phase on IMCL content and use during exercise. Thus, the effect of menstrual cycle phase on lipid metabolism during exercise requires further research.
Effect of diet on substrate selection during endurance exercise
Carbohydrate Loading
Dietary intake can also impact fuel use during endurance exercise. In a series of classic experiments, Hultman, Bergstrom, and colleagues first demonstrated that muscle glycogen levels can be manipulated by diet and exercise. They also observed that following a period of low carbohydrate intake, the resynthesis of muscle glycogen was greater in a leg that had undergone a bout of continuous exercise as compared with a leg that had not exercised, resulting in supercompensated muscle glycogen levels. Subsequently, these investigators determined the effect of consuming a high CHO diet following a bout of glycogen depleting exercise on subsequent muscle glycogen content and exercise performance. A 3-day high CHO diet again resulted in supercompensated muscle glycogen levels and increased time to exhaustion, findings which resulted in a new dietary regime to improve athletic performance. The traditional CHO loading method involved a 6-day taper-diet regime, where on the first 3 days subjects consume moderate CHO (approximately 50% of total calories) and undergo moderate training, followed by three days of high CHO (approximately 70% of total calories) and minimal training. This taper-diet regime can increase muscle glycogen levels by approximately 25% as compared with a normal diet. More recently shorter regimes (1–4 days), with or without prior bouts of glycogen lowering exercise, have been developed and found to increase muscle glycogen stores by 23% to 89%. Thus, there are a multitude of dietary strategies by which muscle glycogen stores can be elevated but they all involve a precompetition phase of high intakes of carbohydrates.
How long muscle glycogen levels remain elevated following supercompensation and what type of CHO is best to supercompensate glycogen stores are important factors for athletes preparing for competition. Following a traditional CHO loading regime (with the initial low CHO phase), CHO levels remain elevated for at least 3 days following the cessation of a high CHO diet and abstinence of physical activity. From a practical standpoint, these findings are important because they imply that athletes can elevate their glycogen stores in the days prior (ie, before having to travel to the event) to a competition and maintain these levels as long as they are not training in the meantime. Whether supercompensated muscle glycogen stores remain elevated beyond 3 days following the cessation of high CHO, and whether other loading regimes also result in glycogen stores that remain elevated for at least 3 days following the cessation of the dietary intervention needs to be determined.
Both simple and complex CHO can be used to supercompensate muscle glycogen stores by 44% to 74%. It is more difficult for women to supercompensate muscle glycogen stores when compared with men: men showed increases in muscle glycogen stores and performance time when consuming a loading protocol with a diet containing 75% CHO and 3 days of reduced intensity tapering of training, but women did not. Other investigators have reported that women do not appear to carbohydrate load with the same magnitude to that seen in men. Subsequent studies found that when the amount of CHO was increased to greater than 8 g/kg per day for both sexes, muscle glycogen content increased for both men and women, yet the women still had a relatively lower magnitude of increase. From a practical perspective, most women will have to increase their energy intake during the loading phase to achieve a CHO intake of greater 8 g/kg per day.
Increased muscle glycogen following a period of CHO loading increases the amount of glycogen available to be used during endurance events and can thus improve performance; however, the effect of CHO loading on performance is controversial, with some studies finding improved performance while others find no change in performance. One study finding no effect of CHO loading on performance in women did not measure muscle glycogen stores and thus, as it is more difficult to increase glycogen stores in women, these subjects may not have had elevated muscle glycogen stores at the onset of exercise and this may explain the lack of effect on performance. The additional studies finding no effect of CHO loading on performance used dietary strategies, where subjects consumed between 61% and 70% CHO, whereas studies finding an effect of CHO loading on performance have used dietary strategies where at least 75% of energy comes from CHO. These findings suggest that at least 75% of energy from CHO is needed in men (and likely women) to improve performance.
Carbohydrate Drinks During Exercise
In addition to CHO loading before an event, athletes can consume CHO during an event in order to increase CHO availability to the muscle to sustain high levels of CHO oxidation. CHO ingestion during endurance exercise appears to spare liver glycogen stores while increasing the reliance on plasma CHO. However, there does not appear to be an effect of CHO ingestion during exercise on muscle glycogen use. Traditionally, glucose, sucrose, or glucose polymers have been consumed, resulting in CHO oxidation rates of 1 gram per minute; however, recent research has found that a combination of glucose and fructose, glucose and sucrose, glucose, fructose and sucrose, and maltodextrin and fructose can increase CHO oxidation rates up to 1.75 grams per minute because they use different transporters. To improve performance, 30 g to 60 g of CHO per hour are required, which translates to approximately 0.5 g/kg to 1 g/kg per hour. Not only can CHO ingestion during exercise extend cycle endurance capacity in events greater than 1 hour, but recent research has shown that CHO ingestion during exercise also increases performance by increasing work output or decreasing the amount of time needed to complete a fixed amount of work, as well as improving performance in events lasting less than 1 hour.
High Fat Diets
Short-term high fat diets can alter substrate use during endurance exercise, increasing lipid oxidation at rest and during exercise. Specifically, high fat diets lasting longer than 3 days increase IMCL content and use, carnitine acyltransferase activity and HSL activity, and decrease pyruvate dehydrogenase activity, thus shifting metabolism to more favorably increase lipid use during exercise. Additionally, several studies, but not all, have found that high fat diets decrease muscle glycogenolysis during exercise. The theory behind increasing fat intake to improve performance is that a high fat intake decreases muscle glycogen stores and increases IMCL content, shifting metabolism toward an increased reliance on fat oxidation. Thus, when muscle glycogen stores are acutely replenished before exercise they are spared, prolonging the ability to maintain a higher VO 2 max. Therefore, the strategy for high fat diets is to increase fat intake for several days before an event, followed by CHO repletion the day before the event to increase fat oxidation and spare glycogen, thus improving performance.
However, the effect of high fat diets on performance is equivocal, with athletes showing an improvement, no change, or a detriment in performance. Additionally, in untrained individuals a period of training while consuming a high fat diet has no effect on or can be detrimental to endurance. Thus, although high fat diets increase lipid oxidation during exercise, they have little effect on athletic performance.
Exercise metabolism and nutrition for inborn errors of metabolism affecting skeletal muscle
The metabolic myopathies are inborn errors of metabolism that impair substrate metabolism in skeletal muscle and result in exercise intolerance ( Table 1 ). Fatty acid oxidation defects (FAOD), glycogen storage disease (GSD), and mitochondrial cytopathies represent the three main groups of disorders. In general, the GSDs present with symptoms during higher intensity and anaerobic type of activities, whereas the FAODs and the mitochondrial cytopthies display symptoms during endurance type activity or under fasted or other metabolically stressful conditions, such as a super-imposed illness. The remainder of this article will consider how exercise training and dietary interventions can be manipulated for individuals with the aforementioned conditions as therapeutic strategies. For a summary of suggested treatments for metabolic myopathies ( Table 2 ).
| Major Category | Examples |
|---|---|
| Mitochondrial myopathy | MELAS, MERRF, CPEO, Kearn-Sayre Syndrome, complex I deficiency, cytochrome b mutations, cytochrome c oxidase mutations. |
| Fat oxidation defect | CPT I, CPT II, TFP, VLCAD, -deficiency; glutaric aciduria type II. |
| Glycogen storage disease | phosphorylase, PFK, acid maltase, phosphorylase b kinase, phosphoglycerate mutase, phosphoglycerate kinase -deficiency. |
| Disease | Treatment |
|---|---|
| Glycogen storage disease |
|
| Fat oxidation defect |
|
| Mitochondrial cytopathy |
|
Glycogen Storage Disease
The glycogen storage diseases refer to a group of disorders characterized by inborn errors of metabolism in glycogen synthesis, glycogenolysis, or glycolysis. Although there are 13 main variants of GSDs with some subcategories, this article shall focus on GSD V (phosphorylase deficiency or McArdle’s disease) and GSD VII (PFK deficiency or Tarui’s disease), given that almost all of the exercise physiology work and dietary manipulation studies have been completed in these GSD subgroups.
Exercise intolerance results from the inability to breakdown glycogen (ie, phosphorylase deficiency) or flux through glycolysis (PFK deficiency) to provide energy via anaerobic or aerobic glycolysis. Although most patients are limited by the impairment in anaerobic glycolysis and manifest symptoms of muscle cramps and pain during the rest-to-exercise transition or with higher exercise intensities, most patients also have a low VO 2 max. The low VO 2 max can be improved by the provision of carbohydrate in the case of McArdle’s disease, but not PFK deficiency. The improvement in exercise capacity and metabolic variables from the infusion of lactate in patients with PFK deficiency further supports the concept that much of the exercise limitation in disorders of glycolysis and glycogenolysis are the result of a lower capacity to provide pyruvate to the tricarboxylic acid cycle via the PDH pathway. Both groups of patients have a hyperdynamic circulatory response to exercise with a rapid increase in heart rate and cardiac output.
Because of the reduced flux through glycolysis, there is a marked attenuation of lactate production during ischemic and semi-ischemic work, and this forms the basis of the forearm exercise diagnostic test. During exercise, P magnetic resonance spectroscopy usually demonstrates a lack of acidosis, with distal defects in glycolysis, such as PFK deficiency, also showing a characteristic increase in phosphomonoesters. With the reduction in glycolytic flux, there is a compensatory increase in flux through the adenylate kinase greater than the myoadenylate deaminase pathway, with the generation of excessive amounts of ammonia and ultimately uric acid, termed “myogenic hyperuricemia.”
For patients with phosphorylase deficiency but not those with PFK deficiency, there is a process called the “second wind phenomenon,” whereby the initial bout of exercise results in tachycardia and breathlessness and muscle cramps. But after a brief rest or a reduction in intensity, there is a reduced perception of effort and the ability to continue exercise at a lower intensity. This phenomenon is caused by the delivery of blood-born substrates (primarily glucose) to the working muscle. From a treatment perspective, most patients adjust their life-style by avoiding precipitating activities, such as brief bursts of high intensity activity. Interestingly, cautious but progressive aerobic conditioning improves functional capacity in patients with McArdle’s disease, occasionally to the lower levels of the healthy control range. Furthermore, McArdle’s disease patients who have a more active lifestyle have a reduced perception of daily muscle pain, and up to 17% can actually attain some level of competitive athletic endeavors.
It is likely that the adaptations to endurance exercise training mentioned above for healthy individuals allows for more rapid substrate delivery and a greater proportion of lipid oxidation at any given exercise intensity. From a nutritional perspective, the consumption of sucrose or glucose 15 to 20 minutes before exercise can provide an exogenous source of glucose and improve exercise capacity in patients with McArdle’s disease. This latter effect is caused by the delivery of glucose to the muscle that can be taken up by GLUT-4 and provide flux through the glycolytic pathway, effectively bypassing the metabolic block.
In contrast, patients with PFK deficiency do worse with glucose ingestion, for it inhibits lipolysis and they cannot take advantage of the exogenous glucose because of the block in glycolysis. Pyridoxine (vitamin B 6 ) supplementation has been suggested for those with McArdle’s disease because it is stored in conjunction with phosphorylase (which is absent) and vitamin B 6 is a cofactor in amino acid oxidation. However, the use of pyridoxine has not been evaluated by a rigorous clinical trial and high doses can cause peripheral neuropathy. High protein diets have also been suggested to up-regulate an alternative fuel pathway, given the intact pathway of amino acid oxidation; however, given that amino acid oxidation represents such a small fraction of total energy supply during exercise and can only be used under aerobic conditions, the clinical utility is questionable.
Along these lines, the acute ingestion of branched chain amino acids did not improve exercise capacity in patients with McArdle’s disease. Given the fact that both PFK and phosphorylase deficiency result in a massive reduction in anaerobic energy transduction, the only degrees of freedom for the cell are the adenylate kinase greater than the AMP deaminase pathway and the phosphocreatine hydrolysis pathway. Consequently, trials have looked at the potential for McArdle’s disease patients to benefit from creatine monohydrate supplementation. One randomized, double-blind trial found that low dose creatine monohydrate supplementation was of some clinical benefit (0.06 g/kg per day); yet a higher dose (0.15 g/kg day) actually impaired exercise capacity. A recent Cochrane review concluded that the only potential therapy for McArdle’s disease was the low dose creatine intervention, with the pre-exercise sucrose being only useful for planned exercise and tending to cause weight gain if sustained regularly for a long period of time.
Fatty Acid Oxidation Defects
The FAODs all result in some impairment of β-oxidation within the mitochondrial matrix. Given the importance of fat oxidation in long duration endurance activity and during periods of fasting (see the section above on fat metabolism and substrate selection during endurance exercise), patients with FAODs generally report symptoms during long-duration endurance activities or when repeated activity is completed in the fasted state or without adequate replacement of carbohydrates. The most commonly documented FAOD leading to rhabdomyolysis and exercise intolerance is CPT II deficiency; however, there are also a wide variety of other FAODs described, including carnitine transporter deficiency, defects of β-oxidation (ie, LCAD, MCAD, trifunctional protein or TFP deficiency), carnitine translocase deficiency, and glutaric aciduria type II (electron transport flavoprotein mutations). Many of these conditions can present in infancy or childhood with a primary liver or encephalopathic picture, while the adult onset forms are predominately myopathic and result in endurance exercise intolerance.
The main myopathic FAODs presenting in adulthood with exercise intolerance include CPT II, TFP, and very long chain acyl-CoA dehydrogenase deficiencies. Although most of the FAODs are autosomal-recessive in their inheritance, it has been reported that more men are diagnosed with CPT II deficiency. This may relate to the previously discussed sex differences in metabolism, where women have a greater ability to oxidize FFA as compared with men, and are somehow protected from the impairment in FFA transport across the mitochondrial membrane seen in CPT II deficiency. Further support for this sex difference in susceptibility to the metabolic effects of CPT II deficiency comes from murine studies showing that male peroxisome proliferator-activated receptor (PPARα −/− ) mice given a blocker of CPT (etomoxir) developed hypoglycemia and died, whereas the females did not.
Many patients with myopathic FAODs are asymptomatic, without the superimposition of a metabolic stress. In contrast to those with glycogen storage disease, these individuals can often perform anaerobic activity for short periods of time without difficulty; however, with more prolonged exercise, patients develop symptoms of cramps, muscle discomfort, and inability to continue the activity. These clinical observations are consistent with metabolic tracer studies in patients with CPT II deficiency, where FFA oxidation is normal at rest and severely impaired during endurance exercise. Standard exercise stress testing is often normal, although the VO 2 max can be somewhat diminished; still, this is neither sensitive nor specific. Occasionally, a high RER during low intensity exercise may indicate the increased reliance upon carbohydrates, although, this finding cannot rule in or rule out an FAOD.
The avoidance of prolonged exercise or exercise during fasting or a superimposed infection is important in the management of those with FAODs. Patients can improve exercise capacity with intravenous glucose infusion, yet do not appear to get the same benefit from oral glucose ingestion. Fortunately, the provision of a high carbohydrate diet (a forme fruste of carbohydrate loading) did lead to improved exercise capacity in those with CPT II deficiency. Although riboflavin to enhance electron transport flavoprotein function, and medium chain triglyceride oil to bypass the carnitine carrier system have been advocated as potential therapies, their efficacy has not yet been demonstrated in a randomized, double blind trial or with metabolic tracers to evaluate the effect on metabolism during exercise. To date, no studies have systematically evaluated the potential benefit of aerobic training in patients with FAODs.
Mitochondrial Disorders
Mitochondrial disorders collectively represent a diverse group of conditions with a primary defect in electron transport chain function. The first mutations definitely linked to a mitochondrial disease were those for Leber’s hereditary optic neuropathy (G11778A), chronic progressive external ophthalmoplegia (mitochondrial DNA deletions) and mitochondrial encepahlopmyopathy, lactic acidosis, and stroke-like episodes (MELAS, A3243G) in the late 1980s. Since then there have been many mitochondrial DNA point mutations described, and more recently a number of nuclear genes encoding for mitochondrial proteins have been discovered. Several reviews regarding mitochondrial medicine and the role of exercise and nutrition have been written recently, and the reader is referred to these for more details.
Many of the cellular consequences of mitochondrial dysfunction can be linked to a decrease in aerobic energy production. Because the mitochondria are the final common pathway for the oxidative metabolism of fats, proteins, and carbohydrates, it is understandable that mitochondrial dysfunction can be precipitated by superimposed metabolic stress, including exercise, infection, or prolonged fasting. Although there is extreme phenotypic and genotypic heterogeneity in the phenotype of the mitochondrial cytopathies, many adult patients will have exercise intolerance as a major feature. The impairment in aerobic energy transduction is reflected by low or very low VO 2 max values. In contrast with the fatty acid oxidation defects and glycogen storage diseases, it is not common for patients with mitochondrial cytopathies to have rhabdomyolysis or myoglobinuria; however, this can be seen in cytochrome b, cytochrome c oxidase, and MELAS A3260G mutations.
Exercise testing in mitochondrial cytopathies usually demonstrates a markedly reduced maximal aerobic capacity (VO 2 max) or a high RER (early lactate production) with cycle ergometry testing (for review, see.) Near infrared spectroscopy or direct venous blood gas measurements often show a lack of deoxygenation consequent to the low oxygen consumption by defective mitochondria. Mitochondrial disorders are a true example of a situation in which the VO 2 max is limited by peripheral extraction and not delivery. Patients with mitochondrial cytopathy show an increase in delivery of oxygen to the periphery, similar to that seen in the glycogen storage diseases. Phosphorus magnetic resonance spectroscopy often reveals an increased reliance on phosphocreatine hydrolysis or a delay in the after-exercise phosphocreatine recovery.
Therapeutic strategies for the mitochondrial cytopathies have focused on increasing aerobic energy transduction through the electron transport chain (cofactors and exercise training), reducing free radical production (antioxidants), bypassing metabolic defects (cofactors, high fat diet), and the provision of alternative energy stores (creatine). Dichloroacetate has been tried to reduce lactate levels by enhancing flux through the PDH pathway, and although lactate is lowered, there were no improvements in function seen in the largest trial to date. Peripheral neuropathy has emerged as a significant side effect with long-term use. The most common cofactor that has been studied is coenzyme Q10, and the balance of the evidence has shown that this compound is safe and has some benefits in surrogate clinical markers of efficacy (for a review of the studies see. ) Coenzyme Q10 has also been used as a therapy for the secondary mitochondrial dysfunction seen in Parkinson’s disease, with some evidence of clinical efficacy and documented improvements in mitochondrial enzyme activity, albeit at doses that were somewhat higher that those traditionally used in mitochondrial cytopathies.
Creatine monohydrate supplementation has been shown to be of some benefit in surrogate markers of strength and endurance and on some clinical features in some, but not all, studies. A recent Cochrane review on the use of creatine monohydrate supplementation did not find that there was a statistically significant improvement in surrogate markers of efficacy when all studies were combined. However, the authors’ finding of higher power output in repetitive high intensity contractions is consistent with the literature in healthy men and women, but of unclear clinical relevance at this point. The authors’ group has found that a combination supplement of creatine monohydrate plus coenzyme Q10 plus α-lipoic acid lowered markers of oxidative stress and lactate in a cohort of subjects with mitochondrial cytopathy in a randomized, double-blind, cross-over study. Given the clinical heterogeneity in the mitochondrial cytopathies and the relatively small numbers of identified patients at most institutions, there are some multicentered trials planned with coenzyme Q10 alone and in combination with creatine monohydrate.
Finally, a diet that is high in fat can theoretically increase the relative contribution of reducing equivalents to complex II of the electron transport chain, and a study has found improved exercise capacity in a cohort of subjects with complex I deficiency given a triglyceride infusion. From a practical perspective, similar benefits were not seen in subjects with complex I deficiency on a high fat diet. Studies have demonstrated improvement in exercise capacity and mitochondrial enzyme activity in subjects with mitochondrial cytopathy following an endurance exercise training program. There was initial concern that endurance exercise may increase the mutational burden and ultimately lead to a worsening of the condition in the long run; however, two subsequent studies did not find evidence for an increased mutational burden following endurance training. Sporadic mitochondrial cytopathies do not result in the accumulation of mutations in the relatively quiescent satellite cells. Consequently, their activation by direct toxin damage and eccentric exercise improves mitochondrial enzyme activity and lowers mutational burden through a process called mtDNA shifting. Larger studies using weight lifting to induce mtDNA shifting will be difficult to complete because of the paucity of cases. However, there may be generic benefits of weight training for patients with mitochondrial disease related to muscle function, similar to those seen in older adults who are known to also have mitochondrial dysfunction, that can be improved with weight training.
This article originally appeared in Neurologic Clinics , Volume 26, Issue 1.
This work was supported by National Sciences and Engineering Research Council of Canada, Canadian Institutes of Health Research, and donations from Warren Lammert and Kathy Corkins and Giant Tiger Stores.
Related posts:
 Recognition and Management of Spinal Cord Injuries in Sports and Recreation
Enhancement Drugs and the Athlete
Neuromuscular Fatigue in Racquet Sports
Neurologic Injuries in Hockey
Head Injuries in Winter Sports: Downhill Skiing, Snowboarding, Sledding, Snowmobiling, Ice Skating and Ice Hockey
Neurologic Running Injuries
Recognition and Management of Spinal Cord Injuries in Sports and Recreation
Enhancement Drugs and the Athlete
Neuromuscular Fatigue in Racquet Sports
Neurologic Injuries in Hockey
Head Injuries in Winter Sports: Downhill Skiing, Snowboarding, Sledding, Snowmobiling, Ice Skating and Ice Hockey
Neurologic Running Injuries
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


