Chapter 52 Multiple Sclerosis
Providers of care to persons with multiple sclerosis (MS) have recently begun to appreciate the value of rehabilitative management techniques because rehabilitation is still the only way to improve function in patients with MS.98
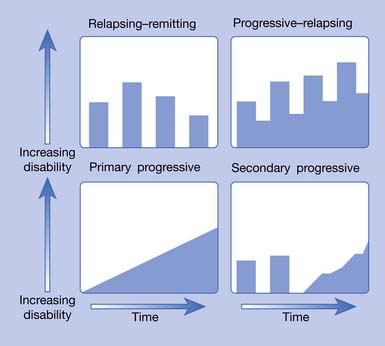
MS is now recognized as a complex disease with at least four pathologic types (Figure 52-1) and four clinical courses (Figure 52-2). As of this printing there are six Food and Drug Administration (FDA)-approved disease-modifying therapies (DMTs) available for the treatment of relapsing–remitting (RR) MS, with more currently in the approval pipeline. All these DMTs are only partially effective in slowing the acquisition of disability. Therefore rehabilitative management continues to play a vital role in the care of MS patients and will continue to do so into the foreseeable future.
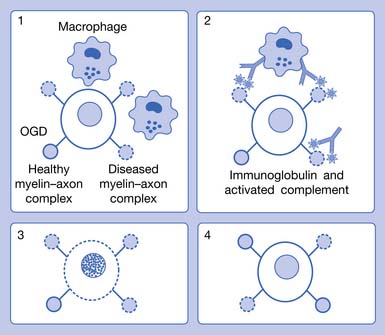
Disease Overview
Demographics
MS is the most common cause of nontraumatic disability affecting young adults in the Northern Hemisphere.62 It is believed there are 400,000 persons in the United States with MS, and the prevalence ranges from 40 to 220/100,000, with the highest prevalence in the highest latitudes, although this differential appears to be lessening.113 Similar latitudinal differences are seen throughout other regions in the Northern Hemisphere. In the Southern Hemisphere, the highest prevalence also appears to be in latitudes farthest from the equator, although the much smaller land mass challenges demographic study.115
In certain populations, such as the Chinese, MS is a relatively rare disease, with current estimates at 20/100,000, even in the northern latitudes of the country (Bo Zhao Chong, personal communication, 2001). In other populations, such as native Africans and Native Americans, MS is also rare. African Americans with MS, however, are prone to a more aggressive course than white Americans.31
Approximately 85% of patients have either the RR or secondary progressive (SP) forms. SPMS typically develops after many years of the RR disease stage. When left untreated, 50% of the RRMS patients will have transitioned into SPMS by 10 years.62 More recent data (probably reflecting the widespread use of DMTs) from Tremlett et al.179 found 58% of RRMS patients converted to SPMS at a mean time of 18.9 years.
The RR/SP form of MS is gender dependent. Currently there are 4 times as many females as males with this form of the disease, a situation showing increasing female preponderance.110,136,196 The typical RRMS patient is a white woman in her late 20s who grew up in a temperate latitude and whose ancestors came from northern Europe.115
Ten to 15% of patients with MS have a disease that is progressive from the onset. This type is called primary progressive (PP) MS and has a roughly equal female to male ratio with a much later onset, around age 40.196 The fourth type of MS, progressive–relapsing (PR), is much less common. These four types are graphically represented in Figure 52-2.
Etiology
Epidemiologic studies have given clues about the etiology of MS. The current view is that it is the product of both a genetic predisposition and an early-acquired unknown environmental trigger, referred to by Kurtzke113 as the “MS affective agent” (MSAA). Migration studies indicate that the likelihood of developing the disease depends on where a person spent the first 15 years of life. Such data suggest that either a causative factor was acquired in the more temperate latitudes or a protective factor was acquired in the less temperate latitudes.36,38,196 Current interest has focused on the low levels of vitamin D—which serves as an immune modulator—in northern latitudes (southern latitudes in the Southern Hemisphere) as a geographic factor.30 Another proposed environmental factor is infection with a variety of candidate viruses (Epstein-Barr is the most frequently mentioned).120,172 A single virus might not be responsible for MS in all patients; different viruses (or virus combinations) might represent the particular MSAA in persons with different, but susceptible, human leukocyte antigen (HLA) types.
The genetic predisposition involves the tissue type. Persons with certain specific tissue (HLA) antigens appear to be either vulnerable to the disease or protected from it. Persons of northern European ancestry, especially those with major histocompatibility complex class II allele HLA-DR2 (HLA-DRB1∗1501) genotype, have a greater chance of developing the disease.6 The expression of this haplotype appears to be regulated by vitamin D, which could explain a link between vitamin D deficiency—a condition which is more common away from the equator—and increased risk for MS.150 The apolipoprotein polymorphism APOE-ε4 is also significantly associated with cognitive impairment in patients with MS.164
An attractive hypothesis regarding the etiology of MS is that genetically susceptible individuals could have an aberration in their immune tolerance, allowing environmental antigens (e.g., viruses) to stimulate production of autoreactive T cells. When such antigens are later encountered in adulthood, they might set off an attack against protein fragments of the person’s own myelin (“molecular mimicry”).112,172,187 This molecular mimicry might later develop into a self-perpetuating degenerative loop through the concept of “epitope spreading.”187 This is one among many postulated mechanisms. Reviews of other hypotheses are available.138
Pathophysiology
The pathologic hallmark of MS is the presence of multifocal demyelinated plaques scattered throughout the central nervous system, with prominent involvement of the periventricular white matter, optic nerves, brain stem, cerebellum, and cervical spinal cord. Demyelination is accompanied by axonal transection and ovoid body formation.177 Another characteristic feature is that these lesions tend to surround the deep veins of the brain, contributing one of the characteristic magnetic resonance imaging (MRI) features of MS, called Dawson’s fingers. These are linear lesions perpendicular to the long axis of the lateral ventricles (Figure 52-3).91
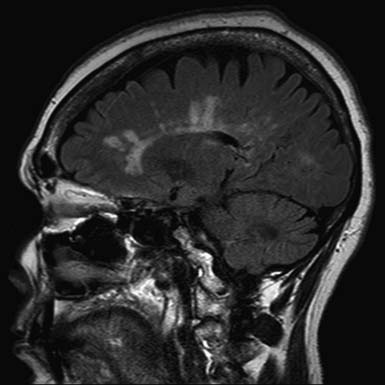
Newer techniques, including magnetic resonance spectroscopy (MRS),111 diffusion-weighted imaging,163 and magnetization transfer ratio (MTR), have shown widespread neuronal deficits in otherwise normal-appearing white matter.188 Progressive development of brain atrophy is a well-known feature of MS. Many studies have shown that atrophy of the brain is present from the earliest stages of the disease and tends to progress as the disease evolves.131 Involvement of gray matter is also evident on dissection but is more difficult to identify with current imaging techniques.34,57,77
For decades investigators have tried to identify a single pathogenic mechanism that would allow the development of more precise immunotherapy. It now appears that subgroups of patients with MS have different pathogenic mechanisms, which might not correspond to their clinical MS types. The results of an international multicenter collaborative study of MS pathology led by Claudia Lucchinetti of the Mayo Clinic have identified four distinct pathologic patterns of MS (see Figure 52-1). Pattern I appears to be purely cell mediated where T cells produce demyelination; pattern II, the most common type, is similar to I, but also appears to involve additional intralesional immunoglobulins and activated complement (cell- and antibody-mediated); pattern III involves apoptosis of oligodendrocytes with selective loss of myelin-associated glycoprotein; and pattern IV involves primary degeneration of oligodendrocytes.122 Patterns I and II target myelin, and patterns III and IV target oligodendrocytes. Differences in antibody recognition and response to immunotherapy have been identified in these four patterns.119
Clinical Presentation
The classic symptoms of MS are manifold and are listed in decreasing frequency in Box 52-1. No other neurologic disease produces the coexistence of so many problems: weakness, fatigue, spasticity, tremor, ataxia, sensory loss, pain, visual loss, cognitive impairment, depression, and neurogenic organ dysfunction (e.g., neurogenic bladder and bowel). The fact that they progress at an unpredictable rate further complicates the management of MS.
Physicians and patients have recently come to realize that cognitive problems and depression occur earlier and much more frequently than previously appreciated. The point prevalence of depression is now thought to be greater than 40%,20 and lifetime cognitive impairment is greater than 60% to 80%.151 Clinicians now know that for every apparent (motor, sensory, or visual) exacerbation, there are as many as 10 (the exact number becomes greater with increasing MRI magnet strength) “subclinical” new gadolinium-enhancing lesions evident on MRI.69 This disease activity can manifest itself by producing emotional or cognitive symptoms. MS was traditionally viewed as a disease of ambulation (indeed, beyond the lower levels, the Kurtzke Expanded Disability Status Scale [EDSS] is a scale of ambulation114 [see Table 52-3]) but is now being viewed also as a disease producing serious “hidden” disabilities (e.g., fatigue, depression, impaired cognition, and pain).109
Neuroimaging
MRI is the most important diagnostic tool in MS. This is reflected in the new McDonald criteria145 (Table 52-1), which allow the diagnosis of MS to be made without a second clinical attack (e.g., new visual, motor, or sensory symptoms) if there is a new lesion on MRI at any point after the initial MRI. A typical MS lesion is ovoid, more than 3 mm in diameter, and located in the periventricular, corpus callosal, or posterior fossa white matter. These can best be seen on fluid attenuation inversion recovery (FLAIR) imaging or T2-weighted MRI sequences (see Figure 52-3). Pathology indicates that such lesions usually correspond to demyelination, although 40% of these—along with “shadow plaques”—contain some degree of remyelination.7 Because of altered physiologic function in partially remyelinated nerves, however, it is questionable whether remyelinated regions again become functional. The increased current leakage, occurring in partially myelinated nerves, results in increased capacitance. This cannot be readily overcome by physiologically generated action potentials; partially demyelinated nerves do not conduct impulses well.90,123,152
Table 52-1 2005 Revised McDonald Criteria for Multiple Sclerosis
| Clinical (Attacks) | Objective Lesions | Additional Requirements to Make Diagnosis |
|---|---|---|
| 2 or more | 2 or more | None; clinical evidence alone will suffice; additional evidence desirable but must be consistent with MS |
| 2 or more | 1 | Dissemination in space by MRI or 2 or more MRI lesions consistent with MS plus positive CSF or await further clinical attack implicating another site |
| 1 | 2 or more | Dissemination in time by MRI or second clinical attack |
| 1 | 1 | |
| 0 (progression from onset) | 1 or more | Disease progression for 1 yr (retrospective or prospective) AND at least 2 of the following: |
Polman C.H., Reingold S.C., Edan G., et al: Diagnostic criteria for multiple sclerosis: 2005 revisions to the McDonald Criteria, Ann Neurol 58:840-846, 2005.
CSF, Cerebrospinal fluid; MRI, magnetic resonance imaging; MS, multiple sclerosis; VEP, visual evoked potential.
McDonald W.I., Compston A., Edan G., et al: Recommended diagnostic criteria for multiple sclerosis: guidelines from the International Panel on the diagnosis of multiple sclerosis, Ann Neurol 50:121-127, 2001.
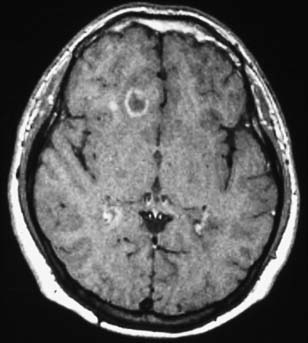
T2 lesions generally persist, whereas 44% of new T1 hypointensities can eventually become isointense and disappear. Enhancing T1 lesions are more reversible than nonenhancing lesions. Ring-enhancing lesions, which can be due to a ring of new inflammation around a previous site of disease, are prone to become persistent T1 “black holes.”157,182 An example of this type of lesion is shown in Figure 52-4. Initial axonal loss and degree of remyelination typically determine the outcome of the lesion.9
Indices of brain atrophy (brain parenchymal fraction and ventricular fraction) show that increased tissue loss is seen in all subtypes of MS. The rate of brain atrophy is higher in younger patients than in older patients.85 Baseline T2 lesion load is a statistically significant predictor of disease-related brain atrophy in patients with established MS.17 A 14-year longitudinal study of a group of patients with “clinically isolated syndrome” (patients who have a symptom that could be MS, but who have not yet developed the second episode in time that is required for the diagnosis) has shown convincingly that baseline T2 lesion load correlates highly with eventual MS diagnosis.11 In this setting, the detection of one or more baseline brain lesions carries a risk of MS of almost 90%. These findings support the use of DMTs in symptomatic patients with MRI abnormalities even before the diagnosis of MS has been established by standard (e.g., McDonald; see Table 52-1) criteria. For those who choose not to take therapy, we recommend having an MRI at least annually to monitor for change in lesion load. Because MS appears to respond best to DMTs in the earliest stages of the disease, there is now an attempt to identify MS as early as possible. Consequently, there is current research on identifying the “radiologically isolated syndrome,” the state before the presentation of the first MS symptoms.143
Other techniques, including those that are now used for research, might soon enter clinical practice. One of these is MRS, which can identify N-acetyl-aspartate (NAA) concentrations. NAA is found in neurons, and a reduction of NAA is a marker for axon damage.111 Another is MTR. These have shown widespread gray matter and neuronal deficits in otherwise normal-appearing white matter.34,77,188
Other than MRI, there are currently no good paraclinical markers to observe patients with established MS. Repeat MRI scans should be obtained when there is uncertainty as to whether a patient is responding well to therapy. These MRIs should include the spinal cord, especially the cervical portion, at least once. Considerable MS activity can occur in the upper spinal cord, but often that area is never imaged. No consensus exists regarding the importance of obtaining routine MRI scans in patients who are clinically doing well on therapy. A case can be made, however, that because MS causes many “hidden disabilities”—such as cognitive impairment—that are less obvious to the observer,109 patients who appear to be “doing well” also might also deserve periodic MRI monitoring, especially if the patient is on submaximal DMT.
Cerebrospinal Fluid Abnormalities
MS remains a clinical diagnosis, but cerebrospinal fluid (CSF) examination can help clarify uncertain cases. White blood cell count and protein levels are normal in two thirds of MS cases; gross elevations of either suggest a different etiology. Immunoglobulin oligoclonal bands are present in 83% to 94% of patients with MS when the test is performed with isoelectric focusing.121 With optic neuritis onset, the presence of oligoclonal bands is also predictive of eventual diagnosis of MS.80
Additional Diagnostic Tests
Evoked potentials (EPs), especially visual evoked potentials (VEP) and somatosensory evoked potentials (SEP), can be used to identify “hidden” lesions, which assist in identifying additional sites of abnormality that are required to make a diagnosis of “multiple” sclerosis. Conduction also demonstrates the state of myelin. Altered or absent myelin causes conduction slowing or conduction block. If conduction slowing through portions of the central neuraxis is found, the diagnosis of a demyelinating disease is strongly supported. Other pathologic conditions that might be mistaken clinically for a demyelinating disease (e.g., vascular lesions or tumor) would likely produce conduction block with attenuation or absence of a response rather than significant conduction slowing.108 Recommendations for the use of SEPs in MS can be found in practice topics published by the American Association of Neuromuscular and Electrodiagnostic Medicine.103
Early studies of VEPs, brain stem auditory evoked responses, and SEPs concluded that VEPs were more sensitive than SEPs in demonstrating central nervous system conduction abnormalities in MS.149,180 Early investigators, however, generally evaluated the short cord SEP pathway available with median nerve testing and did not evaluate SEPs from the lower limbs. Because MS lesions can occur throughout the brain, brain stem, and spinal cord, SEP techniques using the entire central neuraxis are most likely to demonstrate conduction abnormalities. Lower limb SEP testing increases the yield of abnormalities compared with upper limb testing and is comparable with the yield of VEPs.165
VEPs are frequently ordered for the evaluation of a patient with suspected MS to assess the presence of subclinical optic neuritis, in an attempt to confirm the presence of a second site of disease and to confirm that a visual deficit is due to optic neuritis. The technique for these is well established.19 A correlation exists between abnormal VEP latency and MRS-measured NAA levels.71 Optical coherence tomography is an alternative method of assessing the presence of optic neuropathy by measurement of retinal nerve fiber layer thickness.54
Diagnostic Criteria
The revised McDonald criteria (see Table 52-1) represent the current standard of diagnosis.130,145 MRI, CSF, and EP findings can be used to demonstrate dissemination in time and space. In recent years, improvements in MRI techniques have reduced the importance of CSF and EPs in the diagnostic evaluation of MS. The McDonald criteria err on the side of conservancy and might underdiagnose some patients with MS. Examples of this underdiagnosis include those in the early stages of disease or those with primarily spinal cord disease.8,175
For a period after the occurrence of the breakdown in the blood-brain barrier, the affected region is permeable to gadolinium. Consequently, MRI scans taken shortly after an exacerbation (several weeks with a 1.5-tesla magnet; longer with a stronger magnet) after an intravenous injection of gadolinium will show “enhancement” or increased density on T1-weighted images. Such techniques can identify acute lesions, and thereby identify a new lesion in time. It is argued that the observation of both enhancing and nonenhancing lesions confirms the criterion of at least two points in time. Double- and triple-dose gadolinium injections are able to show even more subtle disease activity (seeFigure 52-4).156
Differential Diagnosis
The differential diagnosis for MS is extremely broad and includes metabolic, infectious, vascular, neoplastic, genetic, autoimmune, as well as other central demyelinating disorders (e.g., neuromyelitis optica [NMO] and acute disseminated encephalomyelitis). NMO (Devic disease) is a “demyelinating” disorder consisting of transverse myelitis and optic neuritis that can present similarly to MS. Although similar clinically, it has a different pathogenesis and different treatment, making its differentiation from MS very important. NMO has characteristic features allowing for its differentiation from MS, including longitudinally extensive cord lesions over three or more spinal segments, relatively spared brain MRI, and an associated biomarker, NMO-IgG. NMO-IgG is highly specific (91%) and moderately sensitive (73%) for the diagnosis of NMO.193
Clinically isolated syndrome (CIS) is the term given to the first demyelinating event that might or might not progress to clinically definite MS (CDMS). Typical presentations of CIS include optic neuritis, internuclear ophthalmoplegia, facial sensory loss, sixth nerve palsy, and partial myelopathy. Brain MRI at the time of CIS is predictive of conversion to CDMS with more than 85% of patients with two or more MRI lesions progressing to CDMS, and that risk is greatest in the first 5 years. Brain MRI at presentation of CIS is also predictive of the extent of disability at 10 years. In patients with more than 10 lesions at presentation, 75% show significant functional impairment (EDSS >3 at 10 years), whereas in patients with fewer than 10 lesions at presentation, only 27% demonstrate significant functional impairment (EDSS >3 at 10 years).142
The introduction of the CIS term is clinically relevant as four CIS trials (BENEFIT,86 ETOMS,24 CHAMPS,78 and PRECISE86) have all shown that early treatment with DMT delays progression to CDMS (with a relative risk reduction of about 45% compared with placebo across all studies).
Additional diagnostic tests that can be useful to diagnose MS and the many diseases that can mimic MS can be found in Table 52-2.
Table 52-2 Diagnostic Evaluation for Multiple Sclerosis
| Routine | When Indicated |
|---|---|
| Magnetic resonance imaging: brain and cord | Cerebrospinal fluid studies |
| Antinuclear antibodies | Evoked potentials |
| Lyme titers | HIV |
| Rapid plasma reagin or venereal disease research laboratory test | Electroencephalogram |
| Vitamin B12 | Hypercoagulability studies |
| Thyroid-stimulating hormone | Consultations: rheumatology, psychology, neuroophthalmology |
It should be pointed out that many diseases can mimic MS. Usually (and fortunately) the diagnosis of MS is very clear. But at times, even the most sophisticated MS physician might encounter a disease that appears to be MS but is not. In such cases, Table 52-3 will help sort out the rare imposters from the real disease we are discussing in this chapter.
Table 52-3 Sorting Through the Mimickers of Multiple Sclerosis
| MRI Finding | Disease |
|---|---|
| Brain white matter | |
| Normal | CIS (low risk for MS), NMO (absent or few lesions), ATM |
| Large lesions | AMS (sometimes confluent and perilesional edema), BCS (concentric whorls of alternating rings of enhancement), PACNS (with mass effect) |
| Absent MRI activity at follow-up | ADEM |
| T2-hyperintensity of the temporal pole, U-fibers at the vertex, external capsule and insular regions | CADASIL |
| Diffuse WM involvement | Neuro-Behçet’s disease, HIV encephalitis, small vessel disease, CADASIL |
| Multifocal, asymmetrical lesions starting in a juxtacortical location and progressively enlarging | PML |
| Large lesions with absent or rare mass effect | PML |
| Extensive and bilateral periventricular abnormalities in isolation | Vitamin B12 deficiency, acquired copper deficiency |
| Cortical gray matter | |
| Cortical/subcortical lesions crossing vascular territories | MELAS |
| Prevalent involvement versus white matter | Encephalitis |
| Infiltrating lesions that do not remain in gray or white matter boundaries | Abscesses |
| Deep gray matter | |
| Bilateral lesions | ADEM (at the gray–white matter junction), CADASIL |
| Lacunar infarcts | CADASIL, small-vessel disease |
| T1-hyperintensity of the pulvinar | Fabry’s disease |
| Multiple discrete lesions in the basal ganglia and thalamus | Susac’s syndrome |
| Large and infiltrating basal ganglia lesions | Neuro-Behçet’s disease |
| Infiltrating lesions without respecting gray matter or white matter boundaries | Abscesses |
| T2-hyperintense lesions in the dentate nuclei | AFL (CTX) |
| Spinal cord | |
| Large and swelling lesions | NMO (with corresponding T1 hypointensity), ADEM, ATM, Sjögren’s syndrome |
| Diffuse abnormalities in the posterior columns | Vitamin B12 deficiency, acquired copper deficiency |
| Other | |
| No “occult” changes in the NAWM | NMO, Lyme disease, SID (except in NSLE) |
| Pontine lacunar infarcts | CADASIL, small-vessel disease |
| Dilation of Virchow–Robin spaces | HHC, PACNS |
| Diffuse lactate increase on brain MRS | MELAS |
| Meningeal enhancement | Susac’s syndrome, PACNS, neuro-Behçet’s disease, meningitis, Lyme disease, sarcoidosis |
| Hydrocephalus | Sarcoidosis |
| Absence of optic nerve lesions | PML |
| Regional atrophy | HHC (hippocampus and amygdala), neuro-Behçet’s disease (brain stem) |
ADEM, Acute disseminated encephalomyelitis; AFL (CTX), adult forms of leukoencephalopathy, cerebrotendinous xanthomatosis; AMS, acute Marburg syndrome; ATM, acute transverse myelitis; BCS, Balo’s concentric sclerosis; CADASIL, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy; CIS, clinically isolated syndrome; HHC, hyperhomocysteinaemia; MELAS, mitochondrial myopathy, encephalopathy, lactic acidosis, stroke-like episodes; MRS, magnetic resonance spectroscopy; NAWM, normal appearing white matter; NMO, neuromyelitis optica; NSLE, neuropsychiatric systemic lupus erythematosus; PACNS, primary angiitis of the central nervous system; PML, progressive multifocal leukoencephalopathy; SID, systemic immune-mediated disorders.
Prognosis
A recent natural history study of 2837 MS patients followed prospectively for 22,723 patient years found that progression in MS is slower than previously described. Participants took a median of 27.9 years from onset before requiring a cane. At 15 years after onset, 21% required a cane, increasing to 69% by 40 years. At 30 years after onset, 14% required a wheelchair, increasing to 22% by 40 years. Measured from birth, participants took a median 59 years before requiring a cane. By 50 years of age, 28% required a cane and 6% required a wheelchair. Although men progressed more quickly than women from disease onset, both sexes required a cane at a similar age. This is in contrast to previous studies suggesting that male gender is associated with a worse prognosis. Those with younger age at onset had slower progression but were still significantly younger when requiring a cane compared with those with an older age of onset. This finding also differs from previous reports that younger age of onset is associated with a better prognosis. Male sex and older age at onset were not associated with a worse prognosis in this natural history study. Progression from onset was more rapid in those with a primary progressive course versus those with a relapsing–remitting course. Despite an older age at onset in PPMS, patients were younger when requiring a cane. A primary progressive course was associated with greater progression when measured from both onset and birth. The rate of progression does not differ between PPMS and SPMS once patients reach a level of mild disability. Motor, cerebellar, or brain stem onset symptoms were associated with a more rapid progression from onset compared with sensory or optic neuropathy symptoms; however, patients were of similar age when requiring a cane. Contrary to previous reports, no onset symptom independently predicted a better or worse outcome. Lastly, those progressing rapidly to moderate disability (EDSS 3 in <5 years) required the use of a cane sooner.178
MRI lesion volume at 5 years and the change in volume during the first 5 years correlate with disability scores at 14 years. Early lesion burden might have an important influence on the development of later disability.11
Among the existing measures of clinical status in MS, the most commonly used is Kurtzke’s EDSS, shown in Table 52-4.114 This scale ranges from 0 (no impairment) to 10 (death from MS), with half-point levels along the way. It focuses on mobility more than sensory, bladder, bowel, communicative, or cognitive impairments. In the lower ranges with full ambulation, the so-called functional systems (e.g., sensory, bladder, visual, cognitive) determine the level; in later stages, when ambulation is impaired, gait limitations trump the functional systems. The midrange is heavily weighted on ambulation and vulnerable to interrater and intrarater fluctuations, as well as fluctuations resulting from time of day and other factors. For example, the difference between an EDSS score of 5.5 and 5.0 is the difference in the ability to walk unaided 100 m, but not 200 m. A change in EDSS must be at least two levels (i.e., one full point) to be considered significant because the reliability of half-point changes is poor.119
Table 52-4 Kurtzke’s Expanded Disability Status Scale
| Rating | Description∗ |
|---|---|
| 0.0 | Normal neurologic examination |
| 1.0 | No disability; minimal signs on one functional system |
| 1.5 | No disability; minimal signs on two of seven functional systems |
| 2.0 | Minimal disability in one of seven functional systems |
| 2.5 | Minimal disability in two functional systems |
| 3.0 | Moderate disability in one functional system, or mild disability in three or four functional systems, although fully ambulatory |
| 3.5 | Fully ambulatory but with moderate disability in one functional system and mild disability in one or two functional systems; or moderate disability in two functional systems; or mild disability in five functional systems |
| 4.0 | Fully ambulatory without aid, up and about 12 hr/day, despite relatively severe disability; able to walk without aid 500 m |
| 4.5 | Fully ambulatory without aid; up and about much of day; able to work a full day; might otherwise have some limitations of full activity or require minimal assistance; relatively severe disability; able to walk without aid 300 m |
| 5.0 | Ambulatory without aid for about 200 m; disability impairs full daily activities |
| 5.5 | Ambulatory for 100 m; disability precludes full daily activities |
| 6.0 | Intermittent or unilateral constant assistance (cane, crutch, or brace) required to walk 100 m with or without resting |
| 6.5 | Constant bilateral support (cane, crutch, walker, or braces) required to walk 20 m without resting |
| 7.0 | Unable to walk beyond 5 m even with aid; essentially restricted to wheelchair; wheels self, transfers alone; active in wheelchair about 12 hr/day |
| 7.5 | Unable to take more than a few steps; restricted to wheelchair; might need aid to transfer; wheels self but might require motorized chair for full day’s activities |
| 8.0 | Essentially restricted to bed, chair, or wheelchair, but might be out of bed much of day; retains self-care functions; generally effective use of arms |
| 8.5 | Essentially restricted to bed much of day; some effective use of arms; retains some self-care functions |
| 9.0 | Helpless bed patient; can communicate and eat |
| 9.5 | Unable to communicate effectively or to eat or swallow |
| 10.0 | Death |
∗ Functional systems are the visual, brain stem, pyramidal, cerebellar, sensory, bladder and bowel, and mental systems.
Kurtzke J.F.: Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS), Neurology 33:1444-1452, 1983.
Pregnancy and Breastfeeding
Pregnancy raises several complex issues in MS care. First is the question of the hereditary transmission of MS. An increased incidence of MS exists among the offspring of individuals with MS: 3% for girls and 1% for boys. On the other hand, MS does not appear to have a negative effect on the pregnancy itself or fetal outcome.32
Second is the question of what happens to exacerbation rates during and after pregnancy. During pregnancy, relapse rates decrease to about half of what they would be otherwise.27 During the first 3 months postpartum, the relapse rate is higher than normal. The net effect of pregnancy on the course of MS is neutral, and women do not need to make decisions about pregnancy based on fear that it will worsen their disease.
The next question has to do with management of DMT during pregnancy. Given that the interferon-β drugs are all FDA category C for pregnancy (primarily because of increased rates of miscarriage), and given that the risk of relapse is lower during pregnancy, the standard advice is that interferon-β drugs should be stopped before a woman gets pregnant. Glatiramer acetate is FDA category B for pregnancy and might be a safer choice if a woman wishes to continue DMT during pregnancy. Restarting MS therapy after delivery should be delayed until breastfeeding is discontinued. Breastfeeding might decrease the relapse rate by half during the first 6 months postpartum.67 For protection during this vulnerable period, we recommend 1000 mg of intravenous methylprednisolone monthly during the postpartum nontreatment period, extending several months after treatment has again been restarted.
Medical Management
Drug Therapies
Because MS is an immune-mediated disease, all effective DMTs are immunoactive drugs. These take three forms: DMTs that modify cellular immune responses (immunomodulatory drugs), drugs that interfere with inflammatory cell replication (antiproliferative), and drugs that block extracellular processes. Drugs are also available with multiple actions, including methylprednisolone, which partially protects T-cell migration into the central nervous system and is immunomodulatory at low doses and cytotoxic at high doses. It also reduces edema associated with acute lesions. Table 52-5 outlines the prescribing information for DMT treatments available at the time of the writing of this chapter.
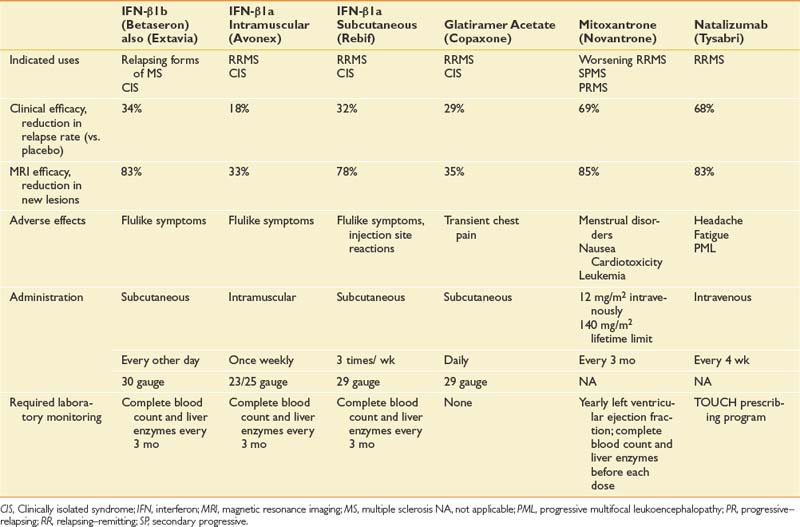
Immunomodulatory Drugs
The initial DMTs were interferon-β. The first was interferon-β1b (Betaseron), which was approved by the FDA for use in RRMS and became available in limited quantities in the United States in 1993.40 This was followed by weekly intramuscular interferon-β1a (Avonex), which gained FDA approval for RRMS in 1996,79 and three times weekly subcutaneous interferon-β1a (Rebif).61,140 Another category of DMT is glatiramer acetate (Copaxone), a polymer of four randomly ordered amino acids (glutamate, lysine, alanine, and tyrosine) in the same molar ratio as in myelin basic protein.81 This is administered subcutaneously daily.
Antiproliferative Drugs
Chemotherapeutic anticancer drugs such as methotrexate, azathioprine, and cyclophosphamide are some of the oldest treatments for MS. These have beneficial results, but the adverse effects limit their use at the most therapeutic dose levels. Data on their effects are limited because it has been difficult to fund large randomized controlled trials with these off-patent medications. More recently, mitoxantrone (Novantrone) has been shown to markedly reduce relapse rate and gadolinium-enhancing MRI lesions in patients with worsening RRMS and SPMS.70 Mitoxantrone has a number of side effects, including menstrual irregularities, nausea, fatigue, and transient leukopenia. The most limiting side effect is that it is cardiotoxic and can cause cardiomyopathy, for which reason the cumulative lifetime dose is limited to 140 mg/m2 (2.5 years with standard dosing).58






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


