Metabolic and Endocrine Abnormalities
Andrew W. Howard
Benjamin A. Alman
INTRODUCTION
Biologic Functions of Bone.
Although orthopaedists tend to focus on the role of bone as the structural support for the body, bone also plays a crucial role in maintaining serum mineral homeostasis. The serum levels of calcium and phosphorus need to be maintained under tight control, to allow for normal function of a variety of cells. The cancellous bone has a tremendously large surface area that allows for the rapid transfer of minerals stored in the bone, such as calcium, to the serum. This process occurs at over a million sites in the human skeleton, mediated by osteoblast and osteoclast cells. A variety of endocrine, metabolic, and cellular factors are crucial to maintain this tight homeostatic balance. Not only do these various factors maintain serum minerals at their proper level, but they also act to regulate the amount of bone present. The interrelationship between these metabolic and endocrine factors with the distribution of minerals between the bone and serum results in metabolic and endocrine disorders altering the quantity and quality of bone. This same interrelationship occasionally results in disorders altering bone structure dysregulating serum mineral balance (1).
Growing Bone.
The effect of metabolic and endocrine disorders on the skeleton is very different in children than in adults. This is because many endocrine and metabolic factors have an effect on the growth plate. Chondrocytes in the growth plate go through a coordinated process of differentiation, beginning with a proliferative phase at the epiphyseal side of the growth plate and progressing to terminal differentiation and apoptotic cell death at the metaphyseal side of the physis. Terminal differentiation is associated with the expression of Type X collagen and the formation of scaffolding for bone formation. Blood vessels located adjacent to the physis in the metaphyseal bone bring pluripotential mesenchymal cells to the region, which differentiate into osteoblasts, producing new bone on the scaffolding left behind by the growth plate chondrocytes. This coordinated differentiation process results in longitudinal growth of long bones. The process of growth plate chondrocyte differentiation needs to be tightly regulated, since if chondrocytes on one side of the body go though this process at a different rate than growth plate chondrocytes on the other side of the body, a limb length inequality would result. The process of growth plate chondrocyte maturation is regulated by both local and systemic factors (2). Conditions in which these systemic factors are dysregulated, as is the case in several endocrinopathies, there is an associated growth plate abnormality (3). In addition, some endocrine factors that regulate bone mineral homeostasis, such as thyroid hormone, also regulate the growth plate chondrocytes. Thus, while thyroid hormone dysregulation has implications in bone density in adults, in growing children, thyroid hormone dysregulation also can cause an abnormality in the growth plate.
FACTORS THAT REGULATE BONE DENSITY
Cells.
Bone density is regulated by osteoblast, osteocyte, and osteoclast cells that add to or break down bone. These cells are regulated by local and systemic factors, some of which can be modulated by the mechanical environment. All of these factors are interrelated, in a complex way that is still not completely elucidated.
Osteoblasts.
Osteoblasts are the main cells responsible for lying down of new bone in the form of osteoid. These cells are
derived from pluripotential stromal precursor cells (sometimes called mesenchymal stem cells) and are the active cells that lay down new bone during skeletal growth and remodeling. Mesenchymal stem cells are very similar too and likely arise from the pericytes or perivascular cells present just deep to the endothelium of blood vessels. A very active area of basic science and translational research is harnessing the regenerative potential of mesenchymal stem cells to treat a variety of diseases (4, 5 and 6). As the bone matures, osteoblasts become encased in the new bone. They produce alkaline phosphatase, an enzyme that is often used to identify osteoblasts and osteoblastic activity. Once they become encased in osteoid, they become relatively quiescent and are termed osteocytes. In mature bone, osteocytes are located extremely far away from neighboring cells, and communicate with other cells through long cytoplasmic processes. The osteocytes remain quiescent until stimulated by hormonal or mechanical factors to begin to reabsorb or lay down bone. Although osteoblasts and osteocytes are thought of as cells responsible for building new bone, they also are able to rapidly reabsorb small quantities of bone. They are able to do this in a relatively rapid manner, in contrast to osteoclasts, which require cellular differentiation and recruitment to reabsorb bone. Thus, they are the first cells that the body activates when bone reabsorption is required (7).
derived from pluripotential stromal precursor cells (sometimes called mesenchymal stem cells) and are the active cells that lay down new bone during skeletal growth and remodeling. Mesenchymal stem cells are very similar too and likely arise from the pericytes or perivascular cells present just deep to the endothelium of blood vessels. A very active area of basic science and translational research is harnessing the regenerative potential of mesenchymal stem cells to treat a variety of diseases (4, 5 and 6). As the bone matures, osteoblasts become encased in the new bone. They produce alkaline phosphatase, an enzyme that is often used to identify osteoblasts and osteoblastic activity. Once they become encased in osteoid, they become relatively quiescent and are termed osteocytes. In mature bone, osteocytes are located extremely far away from neighboring cells, and communicate with other cells through long cytoplasmic processes. The osteocytes remain quiescent until stimulated by hormonal or mechanical factors to begin to reabsorb or lay down bone. Although osteoblasts and osteocytes are thought of as cells responsible for building new bone, they also are able to rapidly reabsorb small quantities of bone. They are able to do this in a relatively rapid manner, in contrast to osteoclasts, which require cellular differentiation and recruitment to reabsorb bone. Thus, they are the first cells that the body activates when bone reabsorption is required (7).
Osteoclasts.
Osteoclasts are derived from circulating monocytes. After differentiation and recruitment to the site of bone where required, osteoclasts are able to reabsorb bone in a very robust manner. They form a ruffled boarder that attaches to the osteoid, in which proteins that degrade the bone matrix are secreted. As such, osteolasts form active reabsorption cavities called Howship lacunae. There is an intimate relationship between osteocyte and osteoclast activities, and many of the signals to activate osteoclasts are mediated by osteocytes. For instance, PTH does not directly regulate osteoclast activity but conveys information via osteocytes, which produce secondary factors that regulate the differentiation of monocytes to osteoclasts. The major signaling pathway that is used by osteocytes to regulate osteoclasts involves a member of the tumor necrosis factor superfamily called RANKL, its receptor, RANK, and a circulating inhibitor, osteoprotegerin (OPG). The receptor, RANK, is present on osteoclast precursor cells, and when stimulated, it causes these precursors to differentiate into active osteoclasts. RANKL is produced by osteocytes that are stimulated to reabsorb bone. OPG also binds to RANKL, but inhibits its ability to activate RANK, and thus inactivates osteoclasts. The balance between OPG and RANKL regulates the number of osteoclasts available (Fig. 6-1) (7, 8). Since OPG inhibits osteoclast production, its use is a promising approach to inhibiting osteoclastic activity, and as such, it has the potential to be developed into useful therapy for osteoporosis, neoplastic bone loss, and even loosening surrounding total joint implants (9, 10). Denosumab, another RANKL inhibitor, has been used in clinical trials to decrease bone turnover and increase bone mineral density (BMD) in postmenopausal females but has not yet been described for clinical use in children (11).
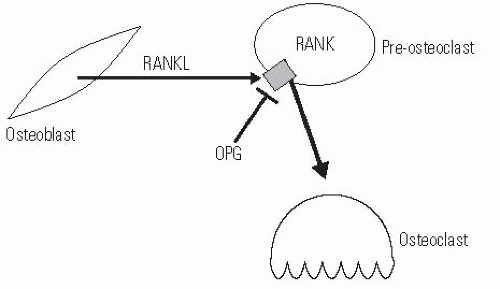 FIGURE 6-1. Expression of RANKL by osteoblasts and osteocytes activates RANK receptor on preosteoclasts to cause differentiation into active osteoclasts. OPG is a circulating factor that can also bind to RANK, but inhibits its ability to cause differentiation to osteoclasts. |
Genetic Mechanisms Controlling Bone Density.
In recent years, there have been tremendous advances made into understanding genes that regulate how these cells develop. Much of this information is covered in several review articles (1, 2) and is beyond the scope of this textbook. For the purpose of this chapter, we consider three modulators of bone density: physical forces, hormone factors, and calcium homeostasis.
Hierarchy in the Regulation of Bone Mass.
There is a hierarchy among the various factors regulating bone mass. Calcium mobilization overrides the other functions of the skeleton. Calcium deficiency due to renal disease, malabsorption, or poor calcium diet invariably causes bone loss, which cannot be overcome by modulating any of the other factors that regulate bone mass. Hormone effects, such as that of estrogen, seem to be more potent than the effect of physical forces. This is suggested by the fact that exercise is limited in its ability to maintain or restore bone mass in postmenopausal women and amenorrhoeic marathon runners lose bone. Of the three modulators of bone mass—calcium availability, hormonal, and physical forces—the last has the least pronounced effects, although this is the one that orthopaedic surgery concentrates most of its efforts on (7).
Calcium Homeostasis
Biologic Functions of Calcium.
Calcium plays a crucial role in the irritability, conductivity, and contractility of smooth and skeletal muscle, and the irritability and conductivity of nerves. Small changes in extracellular and intracellular calcium levels lead to dysfunction of these cells. For the case of neurons, the cellular activity is inversely proportional to the calcium ion concentration, while for cardiac myocytes there is a direct proportionality. Thus, decreases in ionic calcium concentration can lead to tetany, convulsions, or diastolic death. Conversely, increases in the concentration of calcium can lead to muscle weakness, somnolence, and ventricular fibrillation. It is obviously important for the body to guard the concentration of ionized calcium, thus providing a rationale for the overriding importance of calcium homeostasis in modulating bone density (12, 13).
Normal Calcium Balance.
Calcium is absorbed from the gut, stored in bone, and excreted primarily by the kidney. Thus, diseases that effect gut absorption or renal function have the potential to deregulate normal calcium homeostasis, and bone mass. In addition, some conditions that cause massive loss of bone mass, such as widespread metastatic disease or prolonged bed rest, also can alter serum calcium levels. Almost all of the body’s calcium is stored in the bones and is held in the form of hydroxyapatite, a salt that is composed of calcium, phosphorus, hydrogen, and oxygen Ca10(PO4)6(OH)2 in very tiny crystals embedded in the collagen fibers of the cortical and cancellous bone (14, 15, 16 and 17). The small size of the crystals provides an enormous surface area, and this factor, combined with the reactivity of the crystal surface and the hydration shell that surrounds it, allows a rapid exchange process with the extracellular fluid (ECF). This process converts the mechanically solid structure of bone to a highly interactive reservoir for calcium, phosphorus, and a number of other ions (16, 18).
Serum Calcium and Phosphate Naturally Crystallizes.
Hydroxyapatite is not freely soluble in water. At the pH of body fluids, calcium and phosphate concentrations in the serum exceed the critical solubility product, and are predicted to precipitate into a solid form. It is thought that various plasma proteins act to inhibit the precipitation, and keep these ions in solution. This metastable state is important for bone structure, as it allows the deposition of hydroxyapatite during bone formation with a minimal expenditure of energy. Unfortunately, it also makes ectopic calcification and ossification easy to occur as a result of increments in levels of either or both of these ions.
Active Transport of Calcium Regulators.
Calcium cannot passively diffuse across mammalian cell membranes, and as such, requires an active transport machinery to move into or out of cells (12, 16, 18, 19 and 20). Although the mechanism to control this transport is regulated in a large part by the action of the active form of vitamin D, parathyroid hormone (PTH), and the concentration of phosphate (18, 21, 22), a variety of other cell signaling pathways also play a role in calcium transport across cell membranes. These other cell signaling pathways, however, seem to act in specialized cell types under specific physiologic states, and as such, likely play only a small role regulating the total serum calcium level. As such, PTH, vitamin D, and phosphate are the three factors that play the most crucial roles in the calcium transport process, and thus in maintaining the normal extracellular soluble calcium level.
Parathyroid Hormone.
PTH is produced by cells of the parathyroid glands, and the expression level of PTH is regulated by the serum level of ionized calcium. When serum calcium levels are low, there is an increase in PTH expression, protein production, and ultimately increased PTH levels in the serum. There are four parathyroid glands, and any one gland has the potential to produce enough PTH to maintain calcium homeostasis. This is of importance in the surgical management of thyroid neoplasia, in which it is preferable to maintain the viability of at least one parathyroid gland. PTH binds to a family of cell membrane receptors (parathyroid hormone receptors, PTHR), which activate a number of cell signaling pathways. The pathway studied most in the control of calcium is one that regulates adenyl cyclase activity, resulting in an increased cellular level of cyclic adenosine monophosphate (cAMP). cAMP renders the cell membrane more permeable to ionic calcium, and it induces the mitochondria, which are intracellular storehouses for calcium, to release their calcium. These actions increase the intracellular concentration of calcium, but do not promote transport to the extracellular space, a function that also requires vitamin D. PTH acts with 1,25-dihydroxyvitamin D to facilitate cellular calcium transport in the gut, the renal tubule, and in the lysis of hydroxyapatite crystal (20, 21, 23). PTH directly stimulates osteoblasts to begin to degrade the surrounding calcium-rich osteoid. Osteoclasts do not contain receptors for PTH, but are stimulated by PTH activation in osteoblasts through induction of the expression of RANKL, which activates osteoclasts (23, 24). Another action of PTH is to diminish the tubular reabsorption of phosphate, which causes the renal excretion of phosphate (23, 25, 26).
Vitamin D.
Active vitamin D is produced from provitamins through conversion steps in the skin, liver, and kidney (Fig. 6-2). The provitamins are ingested in animal fats (ergosterol) or synthesized by the liver (7-dehydrocholesterol) (14, 20, 27) and are converted to calciferol and cholecalciferol by ultraviolet light, a process that occurs in the skin. In the absence of ultraviolet light, this conversion cannot occur, explaining the vitamin D deficiency associated with prolonged periods indoors away from ultraviolet light sources, such as in chronically ill individuals, or in people living in extremely cold climates (14, 28). The compounds are then transported to the liver, where they are converted to 25-hydroxyvitamin D by a specific hydrolase (29, 30, 31, 32 and 33). Severe liver disease or drugs that block hydrolase activity will inhibit the production of 25-hydroxyvitamin D, also potentially leading to vitamin D deficiency. The final conversion occurs in the kidney. In the presence of specific hydrolases and a number of biochemical cofactors. 25-hydroxyvitamin D is converted to either 24,25-dihydroxyvitamin D or 1,25-dihydroxyvitamin D. The latter serves as the potent calcium transport promoter (34, 35 and 36). A low serum calcium level and a high PTH level cause conversion to the 1,25 analog, while a high serum calcium level, a higher serum phosphate level, and a low PTH level favor formation of 24,25-dihydroxyvitamin D, which is less potent in activating calcium transport (Fig. 6-3) (34, 37, 38, 39 and 40). Serum phosphate also plays an important role here, as a high concentration of phosphate shunts the 25-hydroxyvitamin D into the 24,25-dihydroxy form. Although the 24,25-dihydroxy form is less active in its effects regulating calcium, it has an important role in growth plate chondrocytes. This crucial role for conversion of vitamin D in the kidney, as well as the kidney’s important role excreting excess calcium and phosphorus, explains the particularly deleterious effect of rena l failure on bone homeostasis, causing
vitamin D deficiency as well as directly deregulating normal calcium excretion. Because of the crucial role of vitamin D in calcium metabolism, the National Academy of Sciences and the American Academy of Pediatrics recommend 200 IU per day of vitamin D (41). This dose will prevent physical signs of vitamin D deficiency and maintain serum 25-hydroxyvitamin D at or above 27.5 nmol/L (11 ng/mL). Many professional bodies and experts are currently advocating for increased intake of vitamin D for healthy children, with credible recommendations ranging from 400 to 1000 IU (42, 43). The generic name of 1,25-dihydroxyvitamin D is calcitriol. Recent studies found that vitamin D also has a variety of extraskeletal effects, including modulating the immune response, and as a chemoprotective agent against certain cancers (42, 43 and 44).
vitamin D deficiency as well as directly deregulating normal calcium excretion. Because of the crucial role of vitamin D in calcium metabolism, the National Academy of Sciences and the American Academy of Pediatrics recommend 200 IU per day of vitamin D (41). This dose will prevent physical signs of vitamin D deficiency and maintain serum 25-hydroxyvitamin D at or above 27.5 nmol/L (11 ng/mL). Many professional bodies and experts are currently advocating for increased intake of vitamin D for healthy children, with credible recommendations ranging from 400 to 1000 IU (42, 43). The generic name of 1,25-dihydroxyvitamin D is calcitriol. Recent studies found that vitamin D also has a variety of extraskeletal effects, including modulating the immune response, and as a chemoprotective agent against certain cancers (42, 43 and 44).
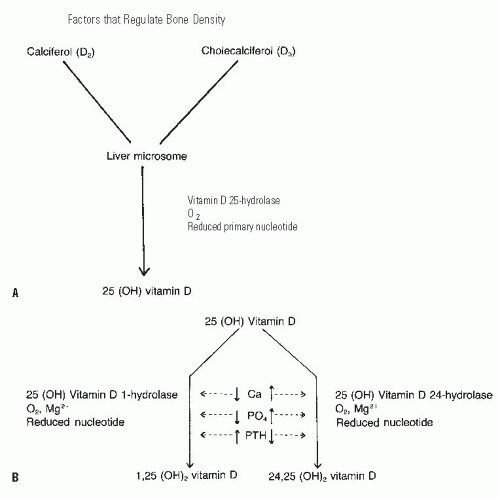 FIGURE 6-2. The conversion of vitamin D from the skin or from dietary sources takes place in the liver and kidney. A: In the liver, the enzyme vitamin D 25-hydrolase acts to form 25-hydroxy vitamin D. B: The second conversion of vitamin D takes place in the kidney, where at least two pathways have been described. The maintenance pathway (when the need is minimal, as defined by a normal calcium and phosphorus and low PTH level) occurs in the presence of a specific enzyme (25-hydroxyvitamin D 24-hydrolase) and results in the less active 24,25-dihydroxyvitamin D. If calcium transport is required, as signaled by the presence of low serum calcium and phosphorus levels and a high PTH level, the body converts the 25-hydroxyvitamine D to the much more active form, 1,25-dihydroxyvitamin D. |
Dietary Calcium Intake.
Dietary calcium is crucial to the maintenance of bone mass. Daily requirements vary with the need of calcium during periods of rapid bone growth. Recommendations from the American Academy of Pediatrics are summarized in Table 6-1 (12, 28, 45, 46). Adequate calcium in the diet during adolescent years is important in the maintenance of bone mass over the long-term, and the orthopaedist should counsel their patients about the importance of appropriate amounts of calcium, as well as vitamin D, in their diet. Several dietary factors alter calcium absorption. Calcium salts are more soluble in acid media, and loss of the normal contribution of acid from the stomach reduces the solubility of the calcium salts and decreases the absorption of the ionized cation. A diet rich in phosphate may decrease the absorption of calcium by binding the cation to HPO42- and precipitating most of the ingested calcium as insoluble material (16, 47). Ionic calcium can be chelated by some organic materials with a high affinity for the element, such as phytate, oxalate, and citrate. Although these materials may remain soluble, they cannot be absorbed (16, 47, 48 and 49). Calcium, in the presence of a free fatty acid, forms an insoluble soap that cannot be absorbed (47, 50). Disorders of the biliary or enteric tracts, associated with steatorrhea, are likely to reduce the absorption of calcium, because it forms an insoluble compound, and because ingested fat-soluble vitamin D is less likely to be absorbed under these circumstances (51).
Dietary Phosphate Intake.
Phosphate (PO4) is absorbed lower in the gastrointestinal tract than calcium and is freely transported across the gut cell to enter the extracellular space, in which it represents a major buffer system. Transport into and out of the bone is passive and related to the kinetics of the formation and breakdown of hydroxyapatite crystals. Tubular reabsorption of phosphate, however, is highly variable, with reabsorption ranging from almost 100% to <50%. The principal factor in decreasing tubular reabsorption of phosphate is PTH.
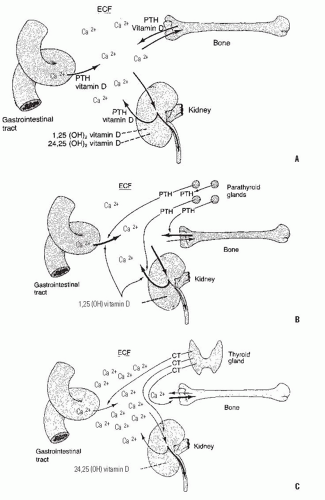 FIGURE 6-3. The roles of the bone, kidneys, gastrointestinal tract, parathyroid gland, and thyroid gland in calcium kinetics. These organs act to maintain calcium in the ECF at the appropriate levels for normal cellular function. A: Vitamin D and PTH act to transport calcium ions across the gut wall and regulate renal excretion and, thereby, bone calcium content. Depending on the need for increased transport, 25-hydroxy vitamin D is converted to 24,25- or 1,25-dihydroxyvitamin D. B: In the normocalcemic state, a reduced concentration of calcium signals the parathyroid glands to release more PTH, which acts at the levels of the gut cell, renal tubule, and bone to increase transport of calcium and rapidly replenish body fluids with it. An increase in PTH also favors the synthesis of 1,25-dihydroxvitamin D in the kidney and acts to promote renal phosphate excretion by markedly diminishing the tubular reabsorption of phosphate. C: In the hypercalcemic state, low concentrations of calcium and PTH act independently to diminish the synthesis of 1,25-dihydroxyvitamin D and decrease transport of calcium in the gut cell, tubule, and bone. Increased concentrations of calcium also cause the release of CT from the C cells of the thyroid gland, thereby diminishing calcium concentration. This mechanism principally involves stabilizing the osteoclast and decreasing its action on the bone, but it is not very effective in humans. |
Endocrine Factors
Sex Steroids.
The most potent endocrine regulator of bone density is estrogen. Much of the clinical and experimental data on the role of estrogen in bone have been generated from data on postmenopausal women. However, clinical data from children with deficiencies in sex hormones, such as in Turner syndrome, show that a lack of estrogen in growing girls also is responsible for profound loss of bone density. The exact mechanism by which estrogen regulates bone formation and loss is unknown. Estrogen receptors are present on both osteoblasts and osteoclasts, yet the cellular mechanism by which estrogen alters these cells’ behavior is not clear. Studies in animals suggest that estrogen exhibits at least some of its effects through the regulation of pluripotential stromal cells in the bone marrow, a process which may be mediated by interleukin-6. Estrogen also suppresses the activation of osteoclasts by inhibiting the activation of RANK in the precursor cells (52, 53).
Androgens also seem to regulate bone mass, although the mechanism is less well understood than for estrogen. Idiopathic hypogonadotropic hypogonadism is associated
with decreased bone mass, and there is an association between delayed puberty and low bone mass in boys, suggesting a positive role for androgens regulating bone mass (54).
with decreased bone mass, and there is an association between delayed puberty and low bone mass in boys, suggesting a positive role for androgens regulating bone mass (54).
TABLE 6-1 Dietary Calcium Requirements | ||||
|---|---|---|---|---|
|
Thyroid Hormones.
Thyroid hormones act in the cell nucleus, interacting with nuclear proteins and DNA to increase the expression of a variety of genes, ultimately positively regulating cell activity. As such, thyroid hormone activates both osteoblasts and osteoclasts. The actual effect on bone mass depends on the body’s balance between these two cell types and how well the normal control of calcium level is able to counteract the heightened activity of these cell types. In general, the balance is in favor of the osteoclast, and most often increased thyroid hormone levels result in bone loss (55, 56).
Corticosteroids.
Corticosteroids have a variety of effects on cells. They inhibit cellular activity in general, potentially decreasing the ability of osteoblasts to lay down new bone. They also have profound effects on the skeleton based on their effect on calcium regulation in the kidney, where they increase calcium excretion. This leads secondarily to elevated PTH levels, with its negative effects on bone density (57, 58).
Calcitonin.
Mechanical Factors.
Excessive reductions in bone strain produced by weightlessness (microgravity in outer space) or immobilization (paralysis, prolonged bed rest, or application of casts) can cause significant bone loss, while strenuous athletic activity can augment certain bones (60, 61). This effect is important in the pediatric orthopaedic population, in which many of the neuromuscular disorders are associated with decreased weight bearing and associated osteoporosis. Bone remodels according to the mechanical stresses applied, a phenomenon termed Wolff law. It is well known that mechanical environment alters cell behavior and gene expression, and it is thought that such a mechanism, most likely acting through osteocytes, is responsible for the effect of weight bearing on bone density as well as for the changes attributable to Wolff’s law (62, 63).
FACTORS THAT REGULATE GROWTH PLATE CHONDROCYTES
In recent years, a number of signaling pathways that regulate the function of growth plate chondrocytes have been elucidated (2). General information about growth plate development and its local regulation is covered in the chapter on developmental biology. However, it is apparent that the growth plate chondrocytes are affected either primarily or secondarily by a variety of endocrine regulatory factors, and as such, a short review here is warranted. Growth plate chondrocytes at the epiphyseal side of the growth plate reside in the resting zone. They begin to proliferate and as such advance toward the metaphyseal side of the growth plate in the proliferative zone. Following this, they enter a prehypertrophic zone, where they shift from proliferation to differentiation. It is also in this prehypertrophic zone that important signals that regulate the differentiation process such as PTH-related protein and Indian hedgehog are present. Following this, the cells hypertrophy form columns in the hypertrophic zone, and then undergo terminal differentiation and cell death. Blood vessels from the metaphysis are present adjacent to the terminally differentiated chondrocytes, bringing in new pluripotential mesenchymal cells, which will differentiate into osteoblasts, forming the new bone on the scaffolding left behind by the chondrocytes. This last region is sometimes called the zone of provisional calcification.
It is easy to imagine how hormones can tip the balance in favor of or against the differentiation process in these cells. In addition, agents that alter normal bone formation by osteoblasts can also alter the growth plate, by preventing the normal replacement of the terminally differentiating chondrocytes with new bone. This inhibition of normal ossification results in the characteristic growth plate changes in rickets, in which there is an increased zone of terminal differentiation. Endocrinopathies can also alter the size and matrix components in the various zones of the growth plate. Such disorders effect terminal differentiation and may make the growth plate mechanically weaker in this region, predisposing to conditions such as slipped capital femoral epiphysis. In a similar manner, it may make the growth plate chondrocytes easier to deform with compressive pressure, causing deformities such as genu varum. This explains the high frequency of these growth plate deformities in children with endocrine disorders. Like in bone, mechanical factors can also play a role in growth pate function. The Hueter-Volkmann principle states that growth plates exhibit increased growth in response to tension and decreased growth in response to compression (64). Thus, an endocrinopathy can cause growth plate deformities, which can then be exacerbated by the effect of the changing mechanical axis in the effected limb.
Similar to the situation in bone, there is also a hierarchal regulation of the growth plate, with endocrine factors playing a dominant role over mechanical factors (3, 65). This is readily apparent in conditions such as rickets, where surgery will not result in correction of genu varum in the absence of correction of the underlying endocrinopathy in the growing child. Thus, it is important to avoid the temptation for surgical correction
of a deformity in a growing child with an endocrine disorder until the endocrinopathy is also treated.
of a deformity in a growing child with an endocrine disorder until the endocrinopathy is also treated.
There are a large number of endocrine factors that play a role regulating growth plate function. In many cases, not much is known about the intracellular signaling mechanisms utilized by these factors. Growth hormone plays an important role regulating growth plate chondrocytes proliferation, mediated by somatomedins. In an absence of growth hormone, there is a slowing of growth plate maturation, as well as a slowing of the rate of long bone growth. Thyroid hormone also plays a role regulating chondrocyte activity, by increasing the metabolic and proliferative rate of the growth plate chondrocytes. PTH may alter growth pate chondrocyte maturation, as the PTH receptor, PTHR1 is expressed in prehypertrophic chondrocytes, and its stimulation results in an inhibition of terminal differentiation. Nutrition and insulin also regulate growth plate chondrocytes, in a similar manner to growth hormone, by regulating growth plate chondrocyte proliferation. A lack of dietary protein exerts a negative control over the somatomedins. Excess glucocorticoids also inhibit growth, partly by an inhibitory effect on protein synthesis in cartilage, but also by interference with somatomedin production and action (3). Although these factors all play roles regulating growth plate chondrocytes, in the coming years we will likely learn more about the role of such factors in a variety of growth plate pathologies, including disorders such as slipped capital femoral epiphysis, where it is well known that a variety of endocrinopathies are predisposing conditions.
DISEASES OF BONE
Rickets
Context/Common Features.
Rickets describes the clinical condition of inadequate mineralization of growing bone. Severe nutritional rickets was endemic in early industrialized societies particularly where sunlight was scarce. Accordingly, severe rachitic deformities were commonly seen in the early days of orthopaedics (66, 67). In developed countries, nutritional rickets is now a rarity, although it may present de novo to pediatric orthopaedists for diagnosis. Inherited form of rickets remain commonly seen in the United States (68). The surgeon should also be familiar with renal tubular abnormalities, which can result in rickets, as well as with the clinical entity of renal osteodystrophy, which describes the bone disease associated with end-stage renal disease and includes features of rickets as well as secondary hyperparathyroidism.
The clinical manifestations of all forms of rickets are similar and, therefore, clinical presentation will be covered separately prior to breaking down the various etiologies.
Clinical Presentation.
Rickets is failure or delay of calcification of newly formed bone at long bone physes. The manifestations include changes in the growth plate morphology with decreased longitudinal growth and angular deformities of the long bones. Osteomalacia, which is failure of mineralization of osteoid formed at cortical and trabecular surfaces, often accompanies rickets in childhood. Osteomalacia is the only result in the adult of the mechanisms, which cause rickets in childhood.
The skeletal abnormalities of severe rickets present in early childhood and often before the age of 2 years. The child may have a history consistent with hypocalcemia in infancy including apneic spells, convulsions, tetany, and stridor prior to age of 6 months (69). The child is often hypotonic with delayed motor milestones for sitting, crawling, and walking. There is proximal muscle weakness and sometimes perfuse sweating. Cardiomyopathy and respiratory and gastrointestinal infections can accompany the clinical presentation (70, 71, 72, 73, 74 and 75).
Skeletal deformities can be evident at every physis. The wrists, elbows, and knees are thickened, and the long bones are short. Genu varum or valgum may be present. Coxa vara may be present. Costochondral enlargement leads to the characteristic rachitic rosary appearance of the chest. Harrison sulcus is an indentation of the lower ribs caused by indrawing against the soft bone. Kyphoscoliosis can be present. Closure of the anterior fontanelle is delayed. Frontal and parietal bossing of the skull is evident. Plagiocephaly may be related to positioning on a soft skull. Delayed primary dentition is common (68, 76).
Radiographic Changes.
The radiographic hallmark of rickets is widened and indistinct growth plates (Fig. 6-4). In a normal child, the distance between the metaphysis and epiphysis of the distal radius should never be >1 mm (77).
Lateral expansion of the growth plates also occurs, particularly with weight bearing. Crawling children weight bear on their wrists, explaining the thickened wrists as well as knees. The metaphysis typically takes on a cupped and splayed appearance. The long bones are short for age. The long bones show evidence of the coxa vara, genu varum, or valgum described above in the clinical deformities. Further evidence of osteomalacia radiographically may also be present. The hallmark is Looser zones. These are transverse bands of unmineralised osteoid, which typically appear in the medial aspect of the proximal femur and at the posterior aspect of the ribs. These are described as pseudofractures and often have an osteosclerotic reaction around them. In an adult, they can progress to true fractures. Acetabular protrusion and pathologic fractures complete the radiographic signs of rickets (70, 76, 77 and 78).
Overview of Classification of Rickets.
Bone is mineralized by the crystallization of calcium and phosphate in the presence of alkaline phosphatase enzyme. Calcium and phosphate are maintained in the body very close to their solubility coefficient by complex series of inhibitors. The control mechanisms in the physiologic state are discussed in sections above.
A useful way of thinking about rickets is to consider those conditions that reduce the availability of calcium, those conditions that reduce the availability of phosphate, and the rare condition that reduces the availability of alkaline phosphatase at the osteoblast-bone junction (Table 6-2). Nutritional rickets and end-organ insensitivity to calcitriol are problems on the calcium side. X-linked hypophosphatemia is the most common form of rickets seen today in the United States and
is caused by renal tubular phosphate wasting in isolation (79). Renal tubular abnormalities including Fanconi syndrome feature renal wasting of phosphate, calcium, magnesium, and bicarbonate. Alkaline phosphatase is deficient only in one rare recessive condition, appropriately called hypophosphatasia.
is caused by renal tubular phosphate wasting in isolation (79). Renal tubular abnormalities including Fanconi syndrome feature renal wasting of phosphate, calcium, magnesium, and bicarbonate. Alkaline phosphatase is deficient only in one rare recessive condition, appropriately called hypophosphatasia.
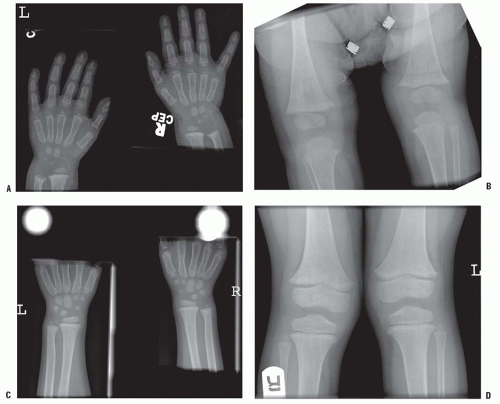 FIGURE 6-4. Rickets. Changes caused by rickets can be seen (A) at the wrist and (B) at the knees of this 1-year-old child with X-linked hypophosphatemia. The growth plates are widened and the metaphyses are cupped, particularly at the ulna and femur. At 4 years of age (C,D), the changes have resolved with medical treatment. |
Finally, renal osteodystrophy is often discussed with rickets and appropriately so since many children with renal osteodystrophy manifest findings of rickets. However, renal osteodystrophy classically includes changes of secondary hyperparathyroidism as well as those of rickets.
Nutritional Rickets.
Nutritional rickets had near universal prevalence in Northern industrialized societies in the 19th century. It has now largely disappeared in developed countries. It remains a significant clinical problem in the developing world with, for example, a 66% prevalence of clinical rickets in preschool children in Tibet in 2001 (80).
The main cause of nutritional rickets is vitamin D deficiency. Vitamin D3 (Cholecalciferol) can be produced in the skin by a process that requires ultraviolet B (UVB) radiation, or it can be ingested in the diet. Peak age of presentation of nutritional rickets is between 3 and 18 months in children who have inadequate exposure to sunlight and no vitamin D supplementation in the diet (81, 82 and 83). Breast milk is poor in vitamin D and prolonged breast-feeding is a risk factor (84, 85). Vitamin D is supplemented in dairy foods in North America and diets deficient in dairy foods are therefore a risk factor (85, 86 and 87). Two hundred international units of vitamin D per day is the recommended dietary amount for preventing rickets (41, 68, 68). Increasing amounts are now being recommended for optimization of bone health, with the Canadian Paediatric Society recommending 800 IU per day for northern children and the AAP recommending 400 IU per day (42). Some experts advocate 1000 IU of vitamin D per
day for all healthy children and adults (43). Sunlight exposure also prevents rickets. Two hours per week of summer sunshine at the latitude of Cincinnati (39 degrees North) is sufficient to produce adequate vitamin D in the skin. However, during the winter months in Edmonton (52 degrees North), there is insufficient UVB exposure to allow for adequate intrinsic production of vitamin D (68). A recent national survey in Canada estimated a prevalence of vitamin D deficiency rickets of at least 3 per 100,000 children, with a higher risk among breastfed children and those dwelling in the north (88).
day for all healthy children and adults (43). Sunlight exposure also prevents rickets. Two hours per week of summer sunshine at the latitude of Cincinnati (39 degrees North) is sufficient to produce adequate vitamin D in the skin. However, during the winter months in Edmonton (52 degrees North), there is insufficient UVB exposure to allow for adequate intrinsic production of vitamin D (68). A recent national survey in Canada estimated a prevalence of vitamin D deficiency rickets of at least 3 per 100,000 children, with a higher risk among breastfed children and those dwelling in the north (88).
TABLE 6-2 Classification of Rickets According to What Is Lacking at the Osteoblast-Bone Interface | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||
Although vitamin D deficiency is the principal cause of nutritional rickets, it is possible to have rickets from a profoundly calcium-deficient diet even in the presence of adequate vitamin D intake (89). Probably much more common is a subtle combination of calcium deficiency and vitamin D deficiency interacting to produce dietary rickets (90). This has been described among the modern Asian population in the United Kingdom (91, 92) and black populations in the United States (68). A diet that is low in calcium and high in phytate, oxalate, or citrate (substances found in almost all fresh and cooked vegetables and that bind calcium) means that calcium intake is poor. Vegetarians, especially those who avoid dairy products, are particularly at risk. This produces an increase in PTH that in turn increases vitamin D catabolism. Vitamin D status may have been marginal due to low sun exposure and poor dietary intake. The increased catabolism of vitamin D with marginal intake results in a vitamin D deficiency and a clinical presentation of rickets. This combination or relative deficiencies of both calcium and vitamin D together has a high prevalence among adolescents presenting with rickets in the United Kingdom and the United States.
Treatment of nutritional rickets involves adequate provision of vitamin D. The treatment dose of 5000 to 10,000 international units per day for 4 to 8 weeks should be provided along with calcium to 500 to 1000 mg per day in the diet (93). Where daily dosing and compliance were a problem, much larger doses of vitamin D (200,000 to 600,000 IU orally or intramuscularly) have given as single doses with good results (94).
Laboratory abnormalities in established nutritional rickets can include a low normal or decreased calcium ion concentration, a low serum phosphate, a low serum 25-hydroxyvitamin D3, and a high alkaline phosphate. Alkaline phosphate drops to normal in response to successful therapy.
Gastrointestinal Rickets.
Even if adequate calcium and vitamin D are present in the diet, some gastrointestinal diseases prevent its appropriate absorption (95). Gluten-sensitive enteropathy, Crohn disease, ulcerative colitis, sarcoidosis, short-gut syndromes have been implicated. If liver disease interferes with the production of bile salts, then fat accumulates in the GI tract and prevents the absorption of fat-soluble vitamins including vitamin D. The vitamin D and calcium deficiency cause bone disease in the same way as nutritional deficiencies, but the treatments are aimed at the underlying gastrointestinal problem as well as at supplementing the missing vitamin and mineral.
X-Linked Hypophosphatemia.
X-linked hypophosphatemia is the most common inherited etiology for rickets with a prevalence of 1 in 20, 000 persons (96). It is an x-linked dominant disorder. This means a female-to-male patient ratio is approximately 2:1, and no male-to-male transmission. Approximately one-third of cases are sporadic (96). People with sporadic occurrence do transmit the defect to their offspring. The defect is in a gene called PHEX (79). This gene product indirectly regulates renal phosphate’s transport. The defect at the kidney is isolated renal phosphate wasting leading to hypophosphatemia. In addition, a low or normal kidney production of 1,25-dihydroxyvitamin D3 is observed, and this would be inappropriate in the hypophosphatemic state.
The clinical presentation includes rickets and mild short stature (97, 98). Dental abscesses occur in childhood, even prior to the development of dental carries (99). Adults with the condition have osteomalacia accompanied by degenerative joint disease, enthesopathies, dental abscesses, and short stature (100, 101 and 102). Specific treatment for the condition is oral administration of phosphate as well as the active form of vitamin D3, calcitriol, which is 1 alpha-hydroxylated. Treatment requires careful metabolic monitoring. Hyperparathyroidism, soft-tissue calcification, and death due to vitamin D intoxication have been problems with medical therapy in the past. Calcitriol can be used in much lower doses than the less active vitamin D metabolites previously used and are thought to be a safer therapy (79, 103, 104). Angular deformities, particularly genu valgum, may persist after medical treatment and require osteotomy (105, 106 and 107). Although good initial corrections are obtained with standard techniques including external fixators, Petje reported a 90% recurrence of deformity after the first surgery and a 60% recurrence of deformity after the second surgery due to ongoing disease activity (108).
A small number of those patients with McCune-Albright syndrome also develop hypophosphatemic rickets. This syndrome includes patients with café au lait spots, precocious puberty, and fibrous dysplasia of multiple long bones. This syndrome is caused by constitutional activation of the cyclic AMP-PKA signaling pathway related to genetic defects in G signaling proteins (109).
1 Alpha-Hydroxylase Deficiency.
In 1961, Prader described what was initially called vitamin D-dependent rickets (110). This was because the initial patients were treated with very large doses of vitamin D. It turns out that these patients have 1 alpha-hydroxylase deficiency and they can be treated with much smaller quantities of the biologically active 1 alpha-hydroxylated calcitriol (111). Typically, the patients present <24 weeks of age with weakness, pneumonia, seizures, bone pain, and the skeletal bone changes of rickets. Serum findings include low calcium and phosphorus, high alkaline phosphatase, and PTH with a normal level of 25-hydroxyvitamin D3, but a markedly decreased level of 1,25-dihydroxyvitamin D3. The patients are not able to convert the accumulated 25-hydroxyvitamin D3 to its biologically active form of 1,25-dihydroxyvitamin D3 and, therefore, develop clinical rickets. The autosomal recessive genetic pattern has been described (112), and the specific mutations were initially described in 1997 (113) since which time at least 31 distinct mutations in the 1 alpha-hydroxylase gene have been identified (111).
Current treatment is oral provision of activated vitamin D3, which is curative.
End-Organ Insensitivity.
In 1978, Marx (114) described two sisters with clinical rickets. The unusual clinical feature was an exceedingly high circulating level of 1,25-dihydroxyvitamin D3. Levels can be 3 to 30 fold higher than normal (115). A striking clinical finding is alopecia or near total loss of hair from the head and the body. These patients have an end-organ insensitivity to vitamin D3 (115). Treatment with very high doses of vitamin D produces a variable but incomplete clinical response. Intravenous high doses of calcium followed by oral calcium supplementation in large quantities have also been tried, but as yet, this rare form of rickets cannot be completely treated medically (111).
Renal Tubular Abnormalities.
There is a large group of causes of the Fanconi syndrome. This syndrome implies failure of tubular reabsorption of many small molecules <50 Da. The kidneys lose phosphate, calcium, magnesium, bicarbonate, sodium, potassium, glucose, uric acid, and small amino acids. With this renal tubular abnormality, there are multiple mechanisms by which bone mineral homeostasis is disrupted (95, 116). As a result, these patients are short with rickets and delayed bone age. The predominant cause of bone disease is hypophosphatemia from renal phosphate wasting, very similar to that seen in x-linked hypophosphatemic rickets. Other mechanisms include calcium and magnesium loss, the metabolic acidosis caused by bicarbonate loss, renal osteodystrophy if renal disease is sufficient that less 1,25-dihydroxyvitamin D3 is produced, and finally decreased calcium and phosphate reabsorption.
Treatment is similar to that of x-linked hypophosphatemia with provision of oral phosphate and vitamin D. Electrolyte imbalances from other causes need monitoring and treatment, and the underlying renal disease can also be treated if possible.
Hypophosphatasia.
This is another disease with clinical overlap with rickets. Hypophosphatasia is caused by alkaline phosphatase deficiency. Like most enzyme deficiencies, this is a recessive condition with over 112 mutations described in the alkaline phosphatase gene in chromosome 1 (117, 118 and 119). Clinically, alkaline phosphatase deficiency produces abnormal mineralization of bone with a presentation of rickets in the child or osteomalacia in the adult (120). Pathologic fractures can occur in children and in adults (121, 122). This is accompanied by abnormal formation of dental cementum that causes loss of teeth. The primary teeth are lost early and with minimal root resorption (123). Additional clinical manifestation can include failure to thrive, increased intercranial pressure, and craniosynostosis.
Hypophosphatasia has an estimated prevalence of 1 per 100,000 people (124). There is a perinatal lethal form. A childhood form presents with rickets at 2 or 3 years of age and remission of the disease in adolescence. An adult form presents with mild osteomalacia with pathologic fractures (117).
There is no satisfactory medical treatment of the underlying defect. Bone marrow transplantation has been used experimentally in severe cases, with the aim of repopulating the bone marrow with osteoblasts capable of producing alkaline phosphatase (117). Surgical treatment of femoral fractures and pseudofractures in the adult has been reported, with rodding techniques superior to plating techniques in the abnormal bone (121).
Renal Osteodystrophy.
Renal osteodystrophy describes the bony changes accompanying end stage renal disease and is commonly seen in patients on dialysis. The clinical presentation includes hyperparathyroidism as well as rickets/osteomalacia in varying combinations (125, 126, 127 and 128).
Renal failure means inadequate clearance of phosphate from the blood once the renal function drops below 25% to 30% of normal. The hyperphosphatemia drives the solubility equilibrium to produce hypocalcemia. This hypocalcemia signals the parathyroid glands to produce PTH, causing secondary hyperparathyroidism. The bony changes of hyperparathyroidism then become evident. These include subperiosteal erosions and brown tumors (Fig. 6-5). The subperiosteal erosions are described as classically appearing on the radial margins of the middle phalanges of digit 2 and digit 3 in adults. In children, they can also be seen at the lateral aspects of the distal radius and ulna and at the medial aspect of the proximal tibia (Fig. 6-6) (129). Prolonged stimulation of the parathyroid glands can produce sufficient hyperplasia that the glands
remain autonomous and maintain a hyperparathyroid state even if the end stage renal disease is treated by transplantation. In this case, the ongoing hyperparathyroidism is described as tertiary rather than secondary.
remain autonomous and maintain a hyperparathyroid state even if the end stage renal disease is treated by transplantation. In this case, the ongoing hyperparathyroidism is described as tertiary rather than secondary.
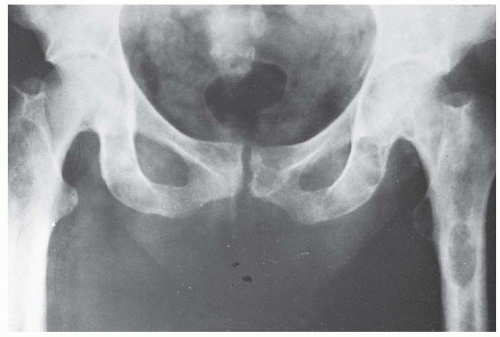 FIGURE 6-5. Radiograph of the pelvis of a patient with renal osteodystrophy shows the marked changes of secondary hyperparathyroidism. Several brown tumors are seen in the femoral shafts and ischial rami. These appear as expanded destructive lesions, resembling primary or metastatic bone tumors. |
The other aspect of renal osteodystrophy is rickets. If there is inadequate renal mass to produce sufficient 1,25-dihydroxyvitamin D3, rickets (clinical and radiographic) will accompany renal osteodystrophy. The clinical manifestations can include varus or valgus deformities at the knees or ankles, with widened and deformed growth plates radiographically, and other radiographic signs of rickets/osteomalacia such as Looser zones (Fig. 6-7).
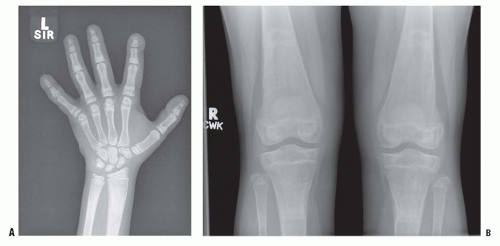 FIGURE 6-6. Renal osteodystrophy in an 8-year-old boy. A: Radiographs of the hand show sclerosis, acroosteolysis, and soft-tissue calcification around the metacarpal phalangeal and proximal interphalangeal joints. B: Radiographs of the knees show subperiosteal resorption at the medial border of the proximal tibia. |
Treatment of renal osteodystrophy includes
Dietary phosphate restriction
Phosphate binding agents, especially those that contain calcium
Vitamin D particularly calcitriol to decrease the secondary hyperparathyroidism as well as to treat clinical rickets or osteomalacia
Restoration of renal function by transplantation often improves the musculoskeletal manifestations.
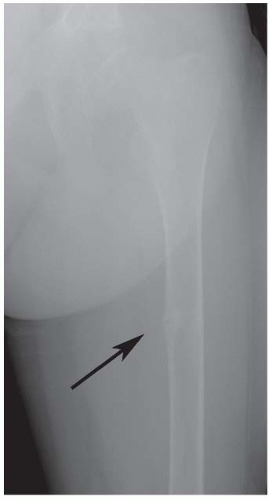 FIGURE 6-7. Renal osteodystrophy. A Looser zone is evident (arrow) in the medial femoral diaphysis. |
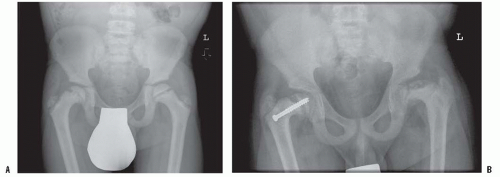 FIGURE 6-8. Renal osteodystrophy in a 12-year-old boy. A: An anteroposterior pelvis x-ray reveals an early capital femoral epiphysis on the right. Slipped capital femoral epiphysis is common in renal osteodystrophy and rare in rickets. B: Three years after fixation the right proximal femoral epiphysis remains open and stable; the left hip now shows signs of epiphyseal avascular necrosis and fragmentation. |
Slipped capital femoral epiphysis occurs frequently in patients with renal osteodystrophy and is not common in other presentations of rickets (Fig. 6-8) (130, 131 and 132). The slip occurs through the metaphyseal side of the physis (126, 133, 134) and occurs at a younger age, in children who are typically small because of their chronic disease. Therefore, stabilization of the slip should permit ongoing growth of the proximal femur if possible. Unstable slips and avascular necrosis are rare in patients with renal osteodystrophy, but avascular necrosis possibly associated with steroid use post transplant has been reported (134). If the child is young and the slip is severe and the bone disease is not yet treated medically, then traction plus medical treatment have shown very good results. When considering the surgical treatment of the slipped epiphysis associated with renal osteodystrophy, the high incidence of bilaterality suggests stabilizing both epiphyses. In young patients, a pinning technique, which allows for growth (smooth pins across the physis), can be considered (133). Hardware cutout, including pin protrusion into the joint, is more likely with the soft bone of renal osteodystrophy but has generally been associated with inadequate medical control of the hyperparathyroidism (133, 134).
Osteoporosis in Children
Implications to General and Lifelong Health.
The National Institutes of Health (NIH) Consensus Panel (2000) has defined osteoporosis as “a skeletal disorder characterized by compromised bone strength predisposing to an increasing risk of fracture.” They note that bone strength includes both bone density and bone quality. Childhood osteoporosis can come from numerous primary and secondary etiologies, summarized in Table 6-3.
There are 10 million people in the United States with osteoporosis, and 18 million more with low bone mass at risk for osteoporosis (135). We associate osteoporosis with senescence, and certainly most of the individuals currently
affected are old, and not likely seeing paediatric orthopaedists. However, the NIH emphasizes that “sub-optimal bone growth in childhood and adolescence is as important as bone loss to the development of osteoporosis.” An epidemiologic study compared rickets mortality in 1942 to 1948 with hip fracture rates in 1986 to 1993 across birth regions in the United States, and found a very high correlation, suggesting that early deficiency of vitamin D could have important effects on the skeleton decades later (136). The recommended intake of calcium for children aged 9 to 17 is 1300 mg per day, and it is estimated that only 10% of girls and 25% of boys meet this minimum requirement (135). While consumption of dairy-based beverages supplying calcium has declined, consumption of carbonated beverages has increased (137). Phosphoric acid is used in cola soft drinks, and teenage girls who drink soft drinks are three to four times more likely to report fractures than those who do not, the association being strongest among active girls drinking cola (138). A meta-analysis of calcium supplementation including 19 randomized trials and 2859 children showed no effect of calcium supplementation alone on BMD at the femoral neck or the lumbar spine (139). Self-reported physical activity in adolescence (but not during adulthood), on the other hand, was a strong determinant of BMD after menopause (140). A meta-analysis of 22 randomized controlled trials of physical activity in childhood showed 1% to 5% increases in bone mineral accrual among the exercising groups, with a greater effect before puberty was complete (141). Vitamin D supplementation has not been so well studied but is receiving increasing attention, with advocates pointing to epidemiologic studies linking vitamin D intake or latitude to lower incidences of cancer and cardiovascular disease as well as to improved bone health (43). A challenge is determining the appropriate level for supplementation of vitamin D, although recent opinion suggests increasing the amount of oral vitamin D3 to 1000 units per day for both children and adults (43).
affected are old, and not likely seeing paediatric orthopaedists. However, the NIH emphasizes that “sub-optimal bone growth in childhood and adolescence is as important as bone loss to the development of osteoporosis.” An epidemiologic study compared rickets mortality in 1942 to 1948 with hip fracture rates in 1986 to 1993 across birth regions in the United States, and found a very high correlation, suggesting that early deficiency of vitamin D could have important effects on the skeleton decades later (136). The recommended intake of calcium for children aged 9 to 17 is 1300 mg per day, and it is estimated that only 10% of girls and 25% of boys meet this minimum requirement (135). While consumption of dairy-based beverages supplying calcium has declined, consumption of carbonated beverages has increased (137). Phosphoric acid is used in cola soft drinks, and teenage girls who drink soft drinks are three to four times more likely to report fractures than those who do not, the association being strongest among active girls drinking cola (138). A meta-analysis of calcium supplementation including 19 randomized trials and 2859 children showed no effect of calcium supplementation alone on BMD at the femoral neck or the lumbar spine (139). Self-reported physical activity in adolescence (but not during adulthood), on the other hand, was a strong determinant of BMD after menopause (140). A meta-analysis of 22 randomized controlled trials of physical activity in childhood showed 1% to 5% increases in bone mineral accrual among the exercising groups, with a greater effect before puberty was complete (141). Vitamin D supplementation has not been so well studied but is receiving increasing attention, with advocates pointing to epidemiologic studies linking vitamin D intake or latitude to lower incidences of cancer and cardiovascular disease as well as to improved bone health (43). A challenge is determining the appropriate level for supplementation of vitamin D, although recent opinion suggests increasing the amount of oral vitamin D3 to 1000 units per day for both children and adults (43).
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


