Fig. 71.1
(a–e) Indolent systemic mastocytosis: radiographs of the right and left knee in a middle-aged female show extensive involvement of the long bones by variable sclerosis (a). The contour of the bones is that of normal modeling excluding a form of skeletal dysplasia. Narrowing of the joint spaces medially is present suggestive of accompanying osteoarthritis. Section from a femoral condyle retrieved at arthroplasty shows thick and somewhat sclerotic bony trabeculae in the subchondral region with a paratrabecular and interstitial cellular infiltrate. Features of osteoarthritis with fibrillation and fissuring of the cartilage and cloning of chondrocytes with reduplication of the tide mark are present (b). The sclerotic bone trabeculae comprise somewhat thick coalescent plates of the bone with somewhat prominent cement lines, giving the bone a somewhat pagetoid architecture (c). Between the trabeculae sheets of cells are noted which have a normal nuclear cytoplasmic ratio. Most have somewhat epithelioid morphology with a tendency to spindling. At higher power, a tendency to spindling of most of the cells is confirmed. There is abundant cytoplasm. Significant cellular atypia is not present. Intermingled eosinophils are prevalent (d). CD117 highlights the abundance of mast cells present in a paratrabecular location (e)
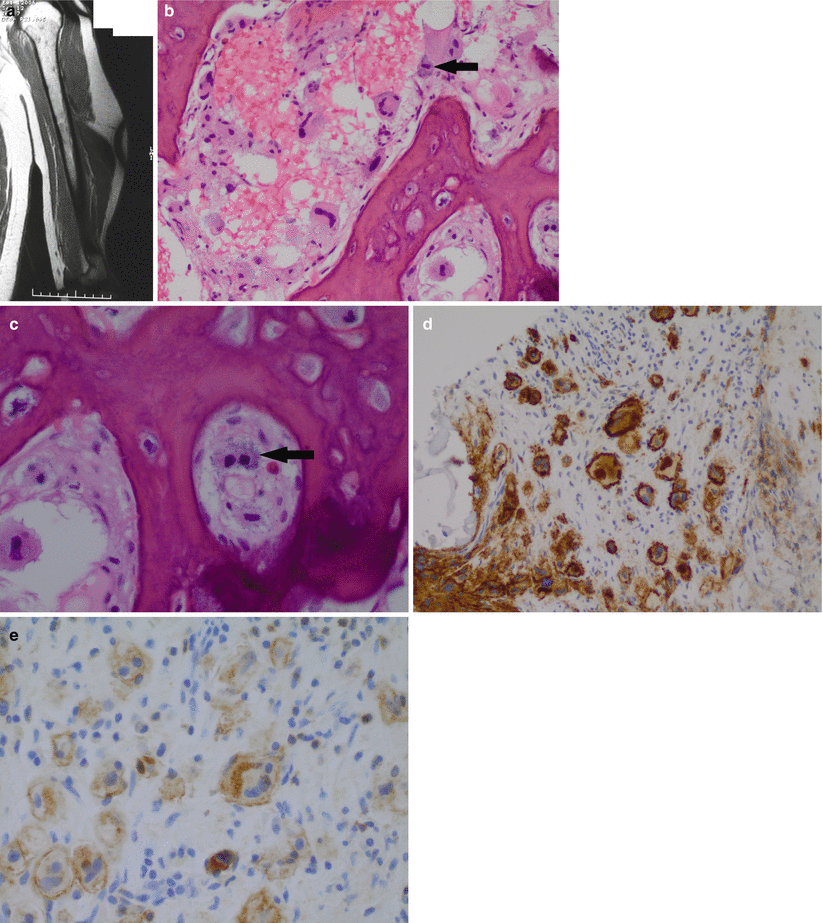
Fig. 71.2
(a–e) Indolent systemic mastocytosis: coronal MR T1-weighted image of a humerus in a male aged 35 with nonspecific arm pain shows subtle abnormality of the marrow with small areas of altered low T1 marrow signal dispersed in a fatty marrow are noted (a). The biopsy shows reactive appearing woven bone with intertrabecular spaces in which enlarged and atypical mast cells are seen including cells with multilobated nuclei and multinucleate forms. Within the cytoplasm, subtle metachromatic granules are visible highlighted with the black arrow (b). At higher power, the metachromatic granules are more easily identifiable in this binucleate cell highlighted with a black arrow (c). The component cells show strong positivity for CD117 (d) and express CD25 (e). The latter allows categorization as systemic mastocytosis. Although the patient had dermatographism, the mast cell infiltration was confined to the bone marrow, and a diagnosis of indolent systemic mastocytosis was made
Systemic mastocytosis with associated hematologic non-mast cell disease (SM-AHNMD)
Aggressive systemic mastocytosis
Mast cell leukemia
Mast cell sarcoma
Extracutaneous mastocytoma
(Smoldering implies a high mast cell burden with intermediate clinical features between indolent and aggressive. Isolated bone marrow mastocytosis occurs without cutaneous involvement. Rare cases of individuals with an accumulation of mast cells with normal morphology and lacking aberrant CD2 and CD25 expression are documented as having “well-differentiated mastocytosis.” This is currently not included in the classification of systemic mastocytosis.)
Synonyms
Mast cell disease
Etiology
The cause is not known.
In most cases, the pathogenesis is linked to activating mutations in the proto-oncogene c-KIT located on chromosome 4q.12 which encodes transmembrane tyrosine kinase receptor.
KIT receptors play an important role in hemopoiesis and are present in mast cells, hemopoietic stem cells, gametocytes, interstitial cells of Cajal, and melanocytes.
Normal mast cell proliferation, differentiation, and activation depend on binding the c-KIT ligand, stem cell factor (SCF), to the KIT receptor.
Abnormalities in SCF regulation or KIT receptors permanently affect the growth, differentiation, apoptosis, and activation of mast cells.
Pathological accumulation of mast cells and tissues ensues.
A variety of mutations are documented.
The commonest is KIT D816V.
(Valine substituted for aspartate at codon 816)
Identified in up to 80 % of cases in adults and children
Similar mutations occur in SM-AHNMD in which the clonal hemopoietic non-mast cell population may exhibit evidence of other genetic events.
Epidemiology
Mastocytosis has been considered rare estimated to occur in between 1 in 1,000 and 1 in 8,000 patients visiting dermatology clinics.
More than 80 % have cutaneous mastocytosis.
The exact frequency of systemic mastocytosis is unknown as indolent forms may elude detection.
A recent population study showed that systemic mastocytosis has a prevalence of 13 per 100,000 population at least in individuals aged over 15 years.
The prevalence increases with age but it is not seen in the very elderly, possibly reflecting less vigorous diagnostic work-up in this age group.
71 % comprise indolent systemic mastocytosis
Of which 2 % were of smoldering form.
22 % had skin lesions alone and likely had associated indolent systemic mastocytosis; however, further investigation was not performed.
Mastocytosis in the bone is rare, affecting less than 200,000 persons.
Sex
No sex predilection is proven.
Age
Mastocytosis can be seen at any age.
Cutaneous mastocytosis is most common in children.
Eighty percent of these present in the first year of life.
Ninety percent are confined to the skin only.
Eighty percent of these undergo spontaneous resolution.
Systemic mastocytosis generally presents in adults after the second decade.
Usually adults with a median age of 54.7 are affected, with a range of 18.6–74.5.
Sites of Involvement
Systemic Mastocytosis
Skin and bone marrow involvement prevail.
Fifty percent of patients with systemic mastocytosis have skin lesions.
In aggressive mastocytosis, involvement of the spleen, lymph nodes, liver, and gastrointestinal tract occurs.
In mast cell leukemia, generalized organ involvement occurs.
Bone involvement occurs in 90 % of cases.
Can be diffuse or focal.
Any bone can be affected.
Predominates in red marrow; thus the axial skeleton, pelvis, and proximal aspects of the long bones are most often affected.
Clinical Signs and Symptoms
These are protean in distribution, severity, and nature.
Ten percent of patients with mastocytosis have systemic disease in addition to skin lesions.
Mast cells are normally widely distributed throughout the body. These cells are sensitive sentinels for the immune system. They detect foreign proteins and initiate a local inflammatory response. This function is made possible by numerous chemical mediators contained in their cytoplasmic granules, among which are histamine, serotonin, and heparin. Many of the local or systemic signs of mastocytosis are due to the release of these substances. These include urticaria, flushing, nausea, and diarrhea.
Children present in infancy or early childhood with one or more macular lesions. They also have pruritus and dermatographia. These skin lesions disappear by late childhood. Systemic mastocytosis, however, is a more extensive disease.
Four main categories of clinical manifestations occur:
General constitutional symptoms
Fatigue, weight loss, fever, and diaphoresis
Cutaneous changes
Nodular eruptions, urticaria, angioedema, and dermatographism.
The most frequent presentation, known as urticaria pigmentosa, is localized mast cell proliferations in the skin of children.
Mediator-related events related to degranulation and release of histamine, tryptase (a major protease), chymase, heparin, platelet-activating factor, prostaglandin D2, cytokines, and chemokines
In the skin
Flushing pruritus, urticaria, and angioedema
Cardiovascular
Hypotension, syncope, and tachycardia
Gastrointestinal tract
Abdominal pain and diarrhea
Respiratory system
Wheezing and throat swelling may occur.
Naso-ocular region
Pruritus
Musculoskeletal system effects
Bone pain
Osteopenia and/or osteosclerosis
Fracture
Arthralgia/myalgia
Dermatographism is seen in all forms.
In aggressive forms, symptoms and signs may reflect organ impairment due to mast cell infiltrates:
Splenomegaly +/− hepatomegaly +/− lymphadenopathy
Hematological findings
Eosinophilia is common.
Anemia.
Leukocytosis.
Neutropenia.
Thrombocytopenia.
Circulating mast cells are seen in mast cell leukemia only.
Objective evidence of mast cell degranulation and mediator release includes:
Increased serum tryptase by at least 20 % + 2 ng/ml above baseline.
In systemic mastocytosis the serum tryptase is >20 ng/ml.
Increased 24 h urinary metabolites of histamine.
Increased 24 h urinary prostaglandin D2 and PDGF2 alpha.
Thirty percent of individuals with SM have associated non-mast cell clonal disorder (SM-AHNMD), and these may present either before or at the time of diagnosis of SM or be unmasked after therapy.
AHMND most often represents myeloid neoplasia especially chronic myeloid leukemia.
Bone Complications and Skeletal Manifestations of Systemic Mastocytosis
Seen in 70–90 % of cases
Often asymptomatic
If symptomatic, usually nonspecific
Pain (28 %) is often poorly localized.
Diffuse osteopenia and osteoporosis is common.
Pathological fracture is seen in 16 %.
Skeletal deformity may occur.
Arthralgia is not infrequent.
Bone modeling is increased.
Bone turnover markers are elevated.
Increased bone turnover appears to correlate with mast cell burden.
Mast cell mediators have a complex effect and bone modeling, and both osteopenia and osteosclerosis are seen, variably distributed and often coexisting.
Rarely, patients with osseous disease lack cutaneous involvement. Patients may present with weight loss, weakness, and hematologic abnormalities due to replacement of the bone marrow.
Image Diagnosis
Changes predominate in the axial skeleton, pelvis, and proximal aspects of the long bones.
Are nonspecific in nature.
Multimodality imaging is most informative.
Changes may vary over time reflecting disease progression and/or therapy.Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree








