mark and will grow disproportionately fast for a few months. The hemangioma is usually a vivid red color with an irregular outline. The lesion does not blanche with direct pressure with the examiner’s finger. These lesions were historically referred to as strawberry naevi or capillary hemangioma. The hemangioma has a natural history that can be divided into three phases: proliferative, involuting, and involuted (10). The proliferative phase is characterized by the rapid dividing of the epithelial cells, whereas the involution phase is slower with far less endothelial activity. There is complete regression of the lesions in 70% of the children by 7 years of age (1). Only 50% of children are left with normal skin; the others may have some residual scaring, telangiectasis, or fibrofatty tissue.
TABLE 9-1 Associations of Vascular Malformations in Children | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||
Slow flow
Capillary malformations (CMs): port wine stains, telangiectasis
Venous malformations (VM)
Lymphatic malformations (LMs): lymphangiomas and cystic hygromas
High flow
Arterial (AMs) and arterial venous malformations (AVMs)
Vascular malformations are more relevant to the orthopaedic surgeon than hemangiomas as they often result in altered limb growth. This can be hypertrophy or atrophy.
Diffuse CM involving an entire limb with congenital hypertrophy of the limb (3). In this condition, the child is born with an enlarged red limb with the adjacent trunk sometimes involved. There is a sharp midline demarcation. The entire limb is enlarged including the soft tissues and bone. There is no progression in the overgrowth after birth. Doppler ultrasound can be used to exclude any AV malformations. No orthopaedic intervention is required.
Diffuse VM of an extremity. The limb is usually blue rather than red (capillary) due to the dermal invasion of the large distorted veins. The venous channels also invade the muscles and the joints, which results in amyotrophy and swelling of the limb. MRI is the best investigation as the T2-weighted images clearly show the soft-tissue and joint involvement (Fig. 9-1). The limb may have slight undergrowth that is usually <2 cm so does not require orthopaedic intervention. Overgrowth is less common and rarely exceeds 2 cm (3). Patients can also get a localized intravascular coagulopathy (LIC) with elevated d-dimer levels and decreased fibrinogen (11). The mainstay of treatment for LIC is low-molecular-weight heparin.
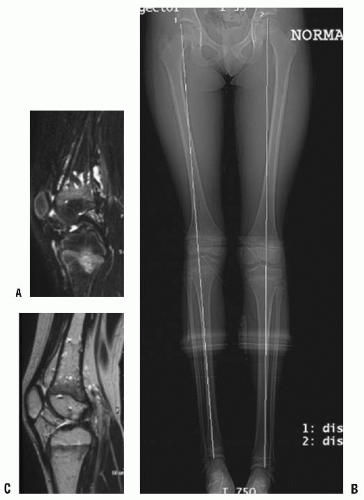 FIGURE 9-1. A-C: Ten-year-old girl with diffuse venous malformation of the right distal thigh with knee joint involvement. The leg is 1.5 cm shorter than the left. |
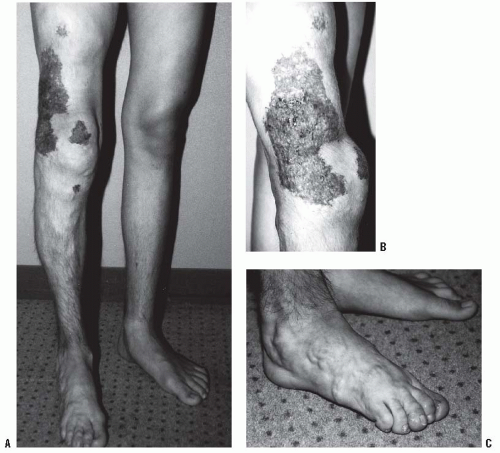 FIGURE 9-2. A: A 15-year-old boy with Klippel-Trenaunay syndrome of his right lower extremities with typical findings of hypertrophy, varicosities, and superficial complex, combined vascular malformations. B,C: He had aching discomfort from the varicosities, intermittent pain from thrombophlebitis, and drainage from the superficial vascular malformations. |
Lindenauer is the only person to document two siblings as having the syndrome (16). Some authors have suggested the cause of Klippel-Trenaunay syndrome is a mutation in the genes that are involved in angiogenesis and vasculogenesis during embryonic development (17, 18, 19 and 20). Tian et al. (21) identified a mutant angiogenic factor (Glu133Lys in VG5Q) in patients with CMs; however, Boon et al. could not find any of these mutations in bona fide Klippel-Trenaunay patients (10). Translocations (5:11, 8:14) have also been reported as being associated with this syndrome (22). Baskerfield et al. studied 33 patients with Klippel-Trenaunay syndrome and, based on extensive vascular studies, concluded that the syndrome was caused by a mesodermal defect. They felt the persisting vascular malformations were fetal microvascular AV communications (17, 18). Unlike hemihypertrophy, children with Klippel-Trenaunay or Parkes-Weber syndrome do not have an increased incidence of Wilms tumor and therefore do not require abdominal ultrasound screening for this (23, 24).
TABLE 9-2 Differences Between Klippel-Trenaunay and Parkes-Weber Syndrome | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
“best guess” the timing of surgical intervention for this variable pattern of discrepancy (38). The exact aetiology of the limb length overgrowth is not known but was originally thought to be caused by venous hypertension (39). A more recent theory suggests there may be some genetic cause for both the vascular abnormalities and alterations in limb length and girth.
with a number of other conditions. It can be confused with Klippel-Trenauany syndrome, epidermal nevus syndrome, neurofibromatosis, idiopathic hypertrophy, Maffucci syndrome, isolated macrodactyly, Ollier disease, and many of the lipomatosis syndromes (53).
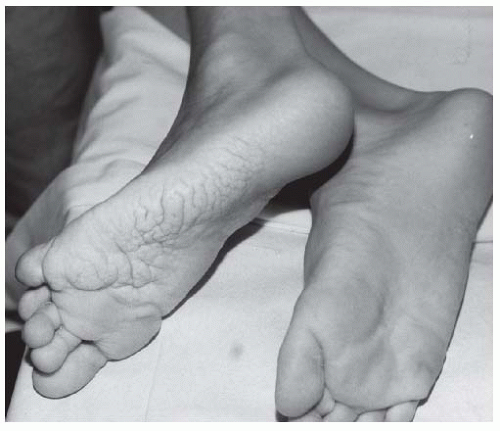 FIGURE 9-3. Adolescent boy with Proteus syndrome with typical gyriform creasing of the sole of his foot. |
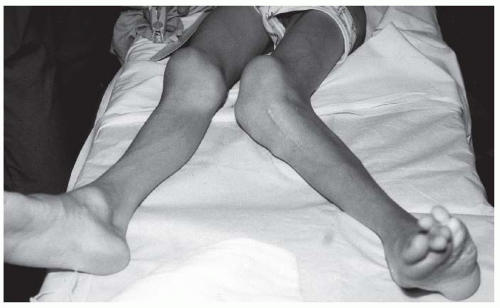 FIGURE 9-4. Adolescent boy with Proteus syndrome with recurrent left genu valgum after a high tibial osteotomy, just prior to repeat osteotomy. |
findings, other causes of primary osteolytic processes must be considered. Torg et al. described a classification system in 1969, which was further expanded by Macpherson et al. in 1973 and remains the most useful one today (94, 95).
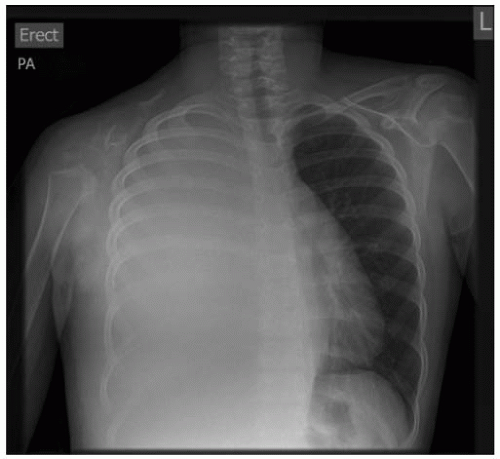 FIGURE 9-5. A 13-year-old girl with Gorham disease who presented with a large right-sided pleural effusion. |
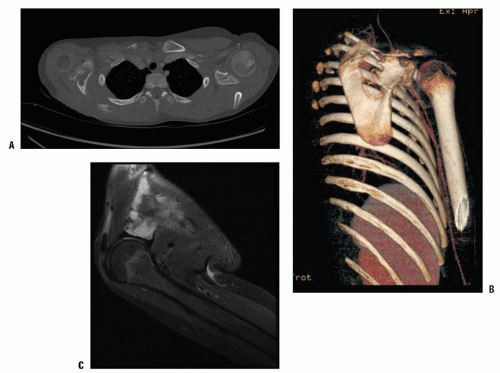 FIGURE 9-6. A 13-year-old girl with CT scan (A,B) showing extensive erosion of the scapula. MRI scan (C) shows the extraosseous soft-tissue extension. |
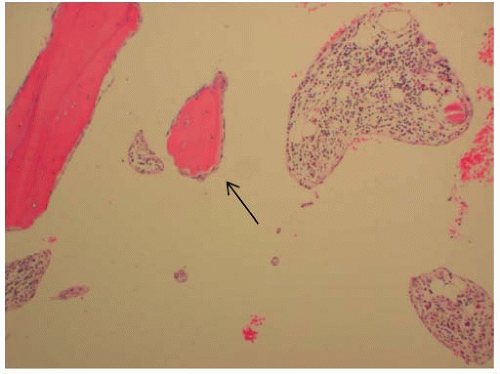 FIGURE 9-7. Histologic slide of a biopsy in Gorham disease showing the thin-walled vessels lined by endothelium cells (arrow) and proteinaceous fluid. |
same problem of asymmetry between the left and right sides of the body that cannot be attributed to normal variation. In this regard, it is important to explain the nomenclature used in addressing these two conditions.
TABLE 9-3 Differential Diagnosis of Hemihypertrophy and Hemihypotrophy | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(sometimes referred to as isolated hemihypertrophy) has no other syndromic features; however, along with Beckwith-Wiedemann syndrome is the only type of hemihypertrophy associated with an increased risk of intra-abdominal tumors (52, 124, 125 and 126).
forms can therefore be categorized according to the area of involvement: classic, segmental, facial, or crossed (121, 122 and 123). Undergrowth may also occur as a result of secondary nonsyndromic hypotrophy, mosaicism for Turner syndrome, Russell-Silver syndrome, neurologic asymmetry (cerebral palsy, polio), osteochondromatosis, endochondromatosis, or polyostotic fibrous dysplasia (144).
proximally the whole limb may be slightly increased in length and width (149, 155, 156). When one sees a macrodactyly of a digit, a very careful clinical examination and sometimes radiologic investigation of the whole limb is paramount to assess the extent of the enlargement. This clinical examination will also help in trying to ascertain whether the macrodactyly is associated with any of the other conditions mentioned earlier. The only radiologic investigation required is usually an x-ray. This is also useful in planning the surgical procedures that may be necessary. The bone age may be advanced in the phalanges involved in the macrodactyly (149, 155).
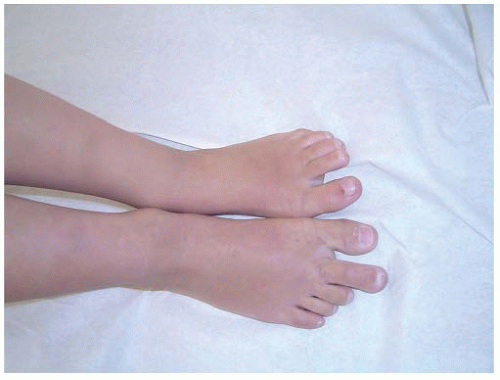 FIGURE 9-8. Macrodactyly of the second toe in a 12-year-old boy. He was asymptomatic and has not required treatment. |
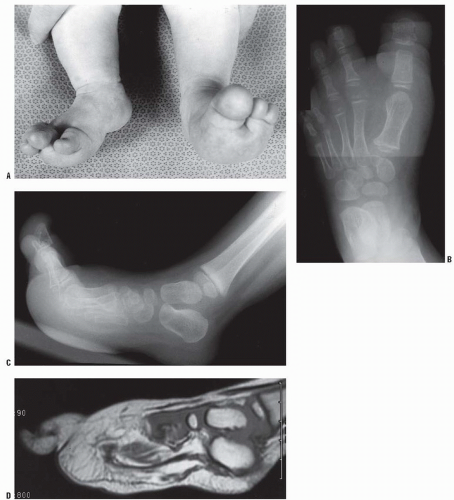 FIGURE 9-9. A 2.5-year-old girl with progressive macrodactyly of both feet, with macrodystrophica-lipomatosa. A: There is significant plantar hypertrophy, resulting in hyperextension of the digits, and there is marked asymmetry in the digital enlargement. B,C: The plain radiographs demonstrate soft-tissue enlargement, as well as underlying bony enlargement. D: The magnetic resonance imaging demonstrates overgrowth of essentially all the elements of the digit, particularly the fibroadipose tissue typically seen in macrodystrophica-lipomatosa. |
children’s fat (152). Usually, there is greater involvement of the tissues distally rather than proximally. There is an increase in the amount of fat and fibrous tissue that surrounds the nerves. The perineurium is thickened and there is proliferation of the fibrous tissue in the endoneurium (149, 151, 152, 157). The muscle is infiltrated in a similar way. The bone is increased in both width and length. Ben-Bassat et al. found that there was a proliferation of fibroblasts or osteoblasts between the periosteum and the cortical bone that may account for the phalangeal overgrowth (149, 155). There does not appear to be any pathologic features in the blood vessels.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


