Chapter 15 Infection and Autoimmunity
Infections are associated with both the etiology and morbidity of systemic lupus erythematosus (SLE). Mounting evidence suggests that viral infections could serve as a trigger for the production of autoantibodies and subsequently lupus. Several viruses have been identified as potential agents, based on association studies, cross-reactivity, or similarity of the symptoms of infection with SLE (Box 15.1). Viruses might trigger the development of autoimmunity through molecular mimicry, alteration of the immune system, the effects of tissue damage, or other mechanisms. After developing lupus, patients are more susceptible to infection with a variety of bacterial, fungal, and viral pathogens. Susceptibility to infection has been associated with both disease activity and the use of immunosuppressive therapies to treat lupus.
BOX 15-1 EVIDENCE SUGGESTING POTENTIAL ROLES FOR INFECTION IN SLE
ETIOLOGY
Twin concordance studies demonstrate the importance, and ironically also the insufficiency, of genetics in lupus causation. Monozygotic twins have a concordance rate for SLE of 25 to 40%, while dizygotic twins have a rate closer to 9%.1–3 The difference between these rates indicates that genetics are indeed important, and genome screening analyses have identified at least 12 chromosomal regions that are linked to SLE.4,5
Autoimmunity through Molecular Mimicry
Many viruses produce proteins with potential epitopes that are very similar to self-antigens targeted in lupus (Table 15.1). Antibodies produced against these proteins have the potential to cross-react with cellular proteins. In the case of lupus, the self-antigens that are recognized by antiviral antibodies are often located in nucleic-acid binding complexes such as the spliceosome, specifically the Sm, and nRNP complexes, other RNA-binding particles such as the Ro/La complexes, and nucleosomes, which contain DNA.6 These cross-reactive antibodies have the potential to initiate an autoimmune response based on improper recognition of self-proteins by antigen-presenting cells.
TABLE 15.1 CROSS-REACTIVITY OF ANTIVIRAL ANTIBODIES WITH SELF-ANTIGENS
| Virus | Viral Antigen | Self-Antigen |
|---|---|---|
| Epstein-Barr virus | EBNA-1 | Sm B/B’ |
| Sm D1 | ||
| Ro | ||
| EBNA-2 | Sm B/B’ | |
| Sm D | ||
| Cytomegalovirus | gB | U1 70K |
| HIV | P24 | Sm B/B’ |
| HRES-1 | P28 | U1 70K |
In a molecular mimicry—based model of autoantibody generation, cross-reactive antipathogen antibodies bind to self-proteins. These opsonized self-antigens can then be phagocytosed by dendritic cells, processed, and presented to T cells in the context of MHC class II, leading to a loss of T-cell tolerance. The presence of viral infection would increase the likelihood of the T cells being stimulated by, rather than tolerized, by antigen presentation (Fig. 15.1). Similarly, B cells could serve as the antigen-presenting cells, but bind the self-antigen through a cross-reactive B-cell antigen receptor instead of soluble antibody. B cells present peptides from their specific antigen with extremely high efficiency,7 and would be highly effective at maintaining and broadening an autoimmune response. Once T-cell tolerance is lost, autoreactive T cells could then provide help for autoreactive B cells, leading to maturation and diversification of the autoantibody response, and eventually culminating in the development of pathogenic autoantibodies and lupus disease.
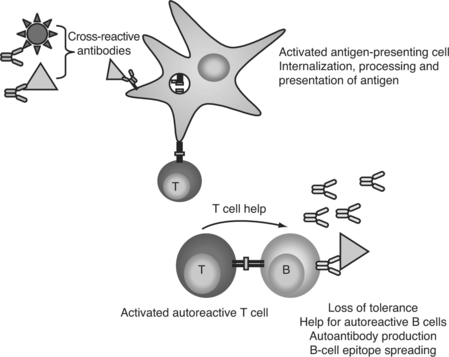
Autoimmunity through Bystander Effects
In the course of antiviral immune activation, some normal safeguards on the immune system are discounted. Virus infection results in immunostimulatory cytokine production. For example, interferon-alpha production is a hallmark of viral infection.8 Lupus patients exhibit elevated levels of interferon-alpha in the blood.9 Microarray analysis of gene expression shows a marked increase in expression of genes that are responsive to interferon-alpha in lupus patients.10–12 Interferon-alpha stimulates dendritic cells to activate T-cell immunity, including autoreactivity.13 Similarly, the presence of viral products activates toll-like receptors on antigen presenting cells,14 causing these cells to mature and present antigen in a stimulatory, rather than tolerogenic, fashion15,16 (Fig. 15.2). Infection could lead to usage of B-variable gene sequences that are prone to react with autoantigens and are not common without the presence of an active immune system.17,18
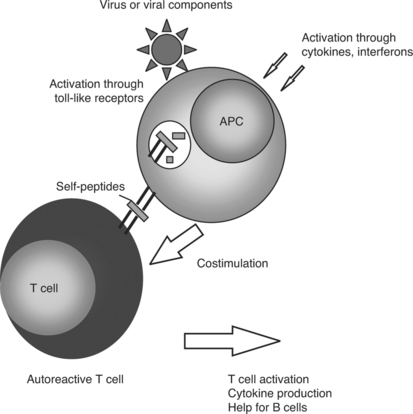
Additionally, viruses fundamentally alter the cells that are infected in ways that would increase their antigenicity. Alterations in apoptosis or clearance of apoptotic cells have been implicated in SLE.19 Viral infections can lead to apoptosis or necrosis due to the cytopathic effect of infection on cells. These mechanisms of cell death alter cellular proteins, potentially unmasking cryptic antigens and making the self-proteins more antigenic.20 Similarly, apoptosis or necrosis leads to the exposure on the cell surface of nuclear antigens, which are the main targets of lupus autoantibodies.21 The increase in cell death because of virus infection may dramatically increase the amount of autoantigen available to activate an inappropriate antibody response. Viral infection can also increase the expression of autoantigens in living cells.22 Similarly, viral infection is controlled by cytotoxic T cells, which kill infected cells via the formation of pores in the cell membrane and the introduction of granzyme B into the target cell. Granzyme B itself cleaves cell proteins, again potentially altering the presentation of self-antigens.23
Desirable Characteristics of Viral Etiologic Agent
Over the past several decades, various infectious agents have been considered in SLE etiology. However, based upon the near impossibility to prove Koch’s postulates with many viral infections due to the lack of animal models and the variability of the outcomes of viral infection, indirect evidence is often required. Several factors contribute to this complexity. First is the multiplicity of factors involved. The process of developing lupus is long, with autoantibodies often appearing years before any clinical signs.24 The virus, if any, involved in SLE pathogenesis, therefore, would not necessarily be present when the symptoms of lupus are noticed. Of course, if the presence of the virus is required to perpetuate or expand the autoimmune response, the virus would be expected to persist for years if not decades.
Epstein-Barr Virus as Etiologic Agent
Epstein-Barr virus is a member of the gamma herpes virus family. This virus primarily infects B cells, and EBV infection in vitro leads to transformation of infected cells. Infection with EBV is lifelong, as the virus is capable of maintaining latency in memory B cells, allowing very little immune surveillance.25 Reactivation from the latent phase occurs periodically.26 The primary viral protein expressed during in vivo latency is Epstein-Barr nuclear antigen 1 (EBNA-1), while the majority of viral genes are expressed during lytic infection.27,28
EBV possesses a multiplicity of characteristics that would make it an ideal initiating or perpetuating agent in SLE (Box 15.1). The lifelong nature of EBV infection, coupled with intermittent reactivation, provides a continual source of potentially cross-reactive antigenic stimulation. In addition, the natural host cells of EBV are B cells, which are responsible for the production of the autoantibodies that are central to SLE. Infection with EBV activates and/or immortalizes B cells, as well as interferes with the normal regulatory mechanisms that control antibody production. In fact, in vitro EBV infection has been used to immortalize B cells producing anti-La and anti-Ro antibodies.29 A protein expressed during viral latency, LMP-1, mimics the signal from CD40,30 which would normally be stimulated by T-cell help. CD40 is a central signal in B-cell activation that provides co-stimulation and allows the B cell to proliferate and undergo antibody isotype class switching and affinity maturation.31 Expression of LMP-1 in mice leads to marked B-cell proliferation and increases in the serum levels of IL-6, suggesting that LMP-1 lowers the threshold for B-cell activation.30 Lowering of this threshold makes a response to inappropriate antigens much more likely.
In addition to the general characteristics of EBV infection, EBV has specific properties that suggest that it may be involved in SLE. Lupus has been associated with alterations in apoptosis, the programmed death of cells. EBV contains a protein, BHRF1, which is homologous to the antiapoptotic protein Bcl-2, and acts to alter the apoptotic pathway in infected cells.32 Similarly, SLE patients demonstrate 7- to 10-fold increases in the levels of interleukin (IL)-10 when compared to controls.33 IL-10 is an immunomodulatory cytokine often expressed in antibody-favoring immune responses. EBV expresses a protein that is highly homologous to IL-10, and acts in a similar manner.34 Additionally, infection of B cells with EBV leads to increased production of IL-10.35 Interestingly, although IL-10 is usually thought of as immunosuppressive, pretreatment of cells with interferon-alpha imbues IL-10 exposure with proinflammatory characteristics.36 Therefore, EBV specifically affects the immune system in ways that are consistent with processes observed to be involved in SLE.
The cross-reactivity of antibodies against EBNA antigens to Ro and Sm antigens is extremely significant in terms of SLE pathogenesis. Investigation into the development of autoantibodies before the onset of symptoms revealed that anti-Ro antibodies are among the first to appear. Anti-Sm antibodies, in addition to being extremely specific for lupus, appear close to diagnosis, suggesting that they are involved in the development of symptoms.34,37
Anti—60-kd Ro autoantibodies, when found alone, develop in a distinct sequence. In patients where the development of anti-Ro antibodies can be observed, the first sequence recognized usually contains amino acids 169 to 180 (TKYKQRNGWSHK). This sequence is cross-reactive with a sequence on EBNA-1 that is bound by antibodies from lupus patients.38 In addition, anti—EBNA-1 antibodies always develop in these patients before anti-Ro antibodies, implicating anti—EBNA-1 immunity in the development of anti-Ro and not vice versa.
Antibodies against the Sm complex are some of the most specific indicators of lupus. Anti-Sm B immunity begins with binding to a single epitope, PPPGMRPP,39 and binding later spreads to encompass multiple epitopes. As is the case with Ro autoimmunity, this initial Sm B’ humoral epitope is highly cross-reactive with a sequence on EBNA-1 that is bound by serum from lupus patients.40 Not only is PPPGMRPP the first antigen bound by the anti-Sm B response, antibodies targeted against this sequence account for an overwhelming proportion of the anti-Sm antibody population, highlighting its critical role in anti-Sm autoimmunity.39
Immunization of animals with this peptide or the corresponding EBNA-1 peptide PPPGRRP leads to widespread autoantibody production and clinical symptoms suggestive of lupus.37,41,42 Similarly, rabbits immunized with the Ro peptide that cross-reacts with EBNA-1, or the corresponding EBNA-1 peptide, developed antibodies to multiple autoantigens, including Sm, nuclear RNP, and dsDNA. These rabbits also developed lupus-like symptoms, including leukopenia, serum creatinine increases, and thrombocytopenia.38 Whole EBNA-1 protein is also able to promote autoantibody generation. DNA vaccination of mice with EBNA-1 coding sequence leads to the development of anti-Sm and anti-dsDNA antibodies.43
During initial infection with EBV, a large panel of both anti-EBV and autoantibodies are generated. Antibodies against the PPPGRRP region of EBNA-1 are transiently present,44 as are lupus anticoagulant antibodies.45,46 Antiphospholipid antibodies, such as lupus anticoagulant, are among the earliest appearing autoantibodies in patients who will develop lupus.24 In normal patients, these antibodies disappear after a short time. However, it may be the case that in patients who will develop lupus, the regulation of the immune system that allows elimination of cross-reactive antibodies is altered, so these antibodies persist.
Case reports of lupus developing subsequent to EBV infection, but not vice versa, detail cases where either the patient is hospitalized for EBV-induced mononucleosis and later develops SLE,47 or patients who are admitted for SLE-related symptoms and found to have indications of EBV infection, such as positive anti-EBV IgM or the presence of EBV antigens in affected tissues.48,49 The timeline of these reports is suggestive, since EBV infection is always determined to have occurred prior to or concomitant with the development of lupus, not the reverse.
Association of EBV with SLE
Because of the attractiveness of EBV as a candidate etiologic agent, attempts to determine if there is an association between EBV infection and SLE has had a long and controversial history. The major obstacles to a clear understanding of the relationship between EBV and SLE are the near ubiquity of EBV infection, the lack of a parallel animal model and the lack of sensitivity of the tests used to detect infection. While seropositivity to EBV approaches 95% in adults in the United States,50 it is only recently that ELISA assays to examine antibody responses and PCR to probe for viral DNA have become sensitive enough to conclusively identify differences in prevalence of infection or to precisely quantify viral or immune differences between patients and controls.
Initial experiments investigating the relationship between SLE and EBV infection focused on differences in antibody titers. The initial studies in this area utilized indirect immunofluorescence to titer anti-EBV nuclear antigen antibodies in two independent studies, and found that lupus patients had significantly higher titers than controls.51
Other studies using immunofluorescence have found no statistically significant difference between antibody titers to EBV between SLE patients and controls. However, of 10 studies examining this question before 1998, six have found significant increases in antibody titer to EBV in lupus patients, one found decreased titers in patients, and three were not able to show a significant difference.52–60
As mentioned, the near ubiquity of EBV infection makes the detection of differences in infection rates difficult. To solve this problem, our group examined sera from a large population of pediatric patients with lupus and carefully matched controls. Because the prevalence of EBV infection is decreased in a younger population, it is possible to accumulate a sufficient number of pediatric samples to obtain adequate power for these studies. This study also used purified antigens as targets for antibody binding by enzyme-liked immunosorben assay (ELISA) instead of immunofluorescence. The results were that 99% of lupus patients were positive for EBV VCA antibodies, while only 70% of controls were positive.61 The increased prevalence of EBV infection in lupus patients was highly significant, with an odds ratio (OR) near 50.61 A second group of pediatric lupus patients was examined in a similar manner. In this cohort, 100% of patients were seropositive for EBV, while 68% of controls had been infected, again a statistically significant result with an OR of more than 14.62
Using these techniques, our group subsequently analyzed a large cohort of adult lupus patients for EBV infection. In this study, 99.5% of patients were seropositive for EBV, while 95% of the controls were positive. Because of the large sample size of 196 patients and 392 matched controls, the study had the power to determine that this result was significant with an OR of 9.35, a 95% confidence interval (CI) from 1.45 to infinity, and a p value of 0.014 when corrected for familial control issues. No significant difference was noticed in seroconversion to HCMV, herpes simplex virus types 1 and 2, or varicella-zoster virus, demonstrating that the differences in EBV infection rate in lupus are specific and not merely the result of nonspecific hypergammaglobulinemia.63
The persistent nature of EBV infection makes it possible to detect EBV DNA in the blood of infected persons years after infection. PCR detection of DNA provides an assay that is not affected by factors such as potential antibody cross-reactivity or non-specific increases in antibody levels. To confirm the results obtained by serologic analysis of pediatric patients, PCR was used to amplify EBV DNA. All 32 of the lupus patients tested positive, while only 23 of the 32 matched controls had detectable EBV DNA in their peripheral blood (OR>10, 95% CI=2.53—infinity, p<0.002).61 A similar study by another group also revealed that a significantly greater proportion of patients tested positive for EBV DNA than controls (81.6% of patients compared to 48.9% of controls, p<0.0001).64
The combined serology and DNA studies show that nearly 100% of lupus patients have evidence of past infection with EBV, while a significantly smaller number of normal controls have been exposed. This pattern suggests that EBV infection is involved in lupus. The association could be explained by EBV acting as a causal agent, or lupus patients being more susceptible to EBV infection. Longitudinal studies of antibody development reveal that EBV antibodies always predate lupus autoantibodies, strongly suggesting that the second scenario is not correct.38
DIFFERENCES IN BIOLOGY OF INFECTION BETWEEN LUPUS PATIENTS AND CONTROLS
Viral Load
Multiple studies have examined EBV viral loads in lupus patients. Real-time quantitative PCR showed a significant increase in the amount of EBV DNA in PBMCs from lupus patients compared to controls.64 EBV DNA levels in both mouthwash samples and blood have been examined using serial dilution of DNA samples. Although there was no difference in the amount of EBV in mouthwash samples, there was a 15-fold increase in EBV DNA concentration in the blood.65 A similar study found an even greater increase in viral load, with lupus patients exhibiting 30-fold higher levels of EBV DNA than controls in the peripheral blood.66
These studies examined only DNA levels, without being able to determine whether the increase in viral load was due to the presence of a larger number of latently infected cells or similar numbers of infected cells with a few cells undergoing lytic infection. During the lytic cycle, viral DNA replication would greatly increase the overall number of viral genomes, skewing the amount of DNA in the blood. To answer this question, limiting dilution was performed on isolated B cells from peripheral blood, and then quantitative PCR was used to quantify EBV viral load on a per cell basis. Using these techniques, it was found that lupus patients have a 10-fold increase in infected cells over controls. Interestingly, in this study patients who were undergoing lupus flares also had significantly increased numbers of EBV-infected cells than patients whose disease was under control.67
These studies, using very sensitive and quantitative methods, all agree that there is a much higher EBV burden in lupus patients than controls. Increased viral burden is important for many reasons. The presence of more infected cells means more chances for the immune system to encounter viral proteins, which may lead to a stronger or altered response. Increased prevalence of infected cells may similarly keep the immune system at a more activated antiviral state, and a proinflammatory cytokine environment may make immune tolerance easier to break in susceptible individuals. In addition, increased prevalence of infected cells means an increased likelihood that one of the B cells immortalized by EBV infection may be a producer of autoreactive antibodies.
Related posts:
 Assessment of Disease Activity in Systemic Lupus Erythematosus
Assessment of Disease Activity in Systemic Lupus Erythematosus
 The Environment in the Pathogenesis of Systemic Lupus Erythematosus
The Environment in the Pathogenesis of Systemic Lupus Erythematosus
 Antibodies and their Antigenic Targets in the Antiphospholipid Syndrome
Antibodies and their Antigenic Targets in the Antiphospholipid Syndrome
 Biology of Dendritic Cells
Biology of Dendritic Cells
 The Genetics of Lupus
The Genetics of Lupus
 What Do Mouse Models Teach Us about Human Systemic Lupus Erythematosus?
What Do Mouse Models Teach Us about Human Systemic Lupus Erythematosus?
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


