Chapter 21 Immune Complexes in Systemic Lupus Erythematosus
BASIC IMMUNOCHEMISTRY OF IMMUNE COMPLEXES: THE PRECIPITIN CURVE
The immunochemistry of immune complexes has been investigated over many decades.1 The classic precipitin curve demonstrates the importance of antigen/antibody ratios in determining the lattice formed by immune complexes in a typical antigen-antibody interaction. Adding increasing amounts of antigen to a constant amount of antibody demonstrates a curve with three general regions: the zone of antibody excess (“pro-zone”), the zone of equivalence, and the zone of antigen excess (“post-zone”). In some antigen-antibody systems, the pro-zone shows an extended region without precipitation. Immune complexes formed in the zone of far antigen or antibody excess are soluble. Large-lattice immune complexes containing IgG, formed at antigen-antibody ratios close to the zone of equivalence, have multiple IgG Fc-regions available for interaction with C1q complement proteins and therefore activate complement efficiently.
Immune complex lattice structure can be altered if there is interaction with complement proteins, because covalently bound complement peptides sterically inhibit immune complex interaction and extended lattice formation. Once immune precipitates are formed, their size can be reduced, leading to solubilization of preformed immunoprecipitates via activation of the alternative pathway of complement.2 Activation of the classical pathway of complement can inhibit immune complex growth by preventing extended lattice formation.3 Thus, in the presence of complement the precipitin curve can be more properly considered a precipitin “surface” in which either higher concentrations of complement components or antigen or antibody excess lead to smaller immune complexes or reduced immune precipitation (Fig. 21.1). Complement activation thus serves as a negative regulator of immune complex lattice extension.
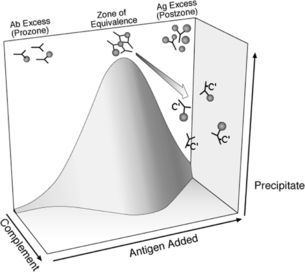
Hypocomplementemic sera from patients with SLE, in comparison with normal sera, fail to prevent formation of immune precipitates. This defective complement-dependent prevention of immune precipitation is seen in early cases of SLE. Prevention of immune precipitation correlates positively with levels of C4A4 and inversely with the present of antibodies to C1q.5
DISEASE ASSOCIATIONS
Immune Complexes and SLE
Active renal SLE is associated with high serum concentrations of antidsDNA antibodies and enrichment of antiDNA within glomerular eluates of patients with SLE, supporting the role of antiDNA in the pathogenesis of SLE. DNA-antiDNA immune complexes are thought to be a central contributor to immune complex nephritis in SLE.6, 7 Several investigators have found evidence for circulating DNA-antiDNA immune complexes and other immune complexes in SLE patients and experimental models.8
Antibodies to the collagen-like region of C1q (anti-C1q) were also found to be concentrated within glomerular basement fragments isolated from kidneys of lupus patients.9 Together with data demonstrating a strong association between lupus nephritis and serum levels of anti-C1q, these data strongly implicate anti-C1q in the pathogenesis of lupus nephritis. Anti-C1q tends to be present if there are multiple autoantibodies (including antidsDNA, antiSSA, antiSm, or others) present and enriched in glomerular basement membrane fragments from kidneys of patients with SLE, suggesting a role for anti-C1q in promoting aggregation of immune complexes in the basement membrane.10
Assays for circulating immune complexes have been used to monitor SLE activity. Numerous studies have suggested that immune complex assays based on C1q binding or C3 content are positive in patients with SLE and can be helpful in assessing disease activity in patients with SLE.8 Assays for antidsDNA and complement components are more widely available than immune complex assays, and are similarly used to monitor disease activity and assist in the diagnosis of SLE. Therefore, measurement of immune complexes is not widely used clinically in comparison with those other measurements.
PATHOPHYSIOLOGIC MECHANISMS
Immune Complexes as Initiators and Regulator of the Autoimmune Response
Immune complexes have traditionally been thought of as having a critical role in causing tissue damage associated with SLE. The potential importance of immune complexes to initiate and enhance the immune dysregulation observed in SLE is also recognized. Immune complexes play a central role by harnessing the specificity of the acquired immune system (high-affinity autoantibodies) to augment the potent but less specific inflammatory response of the innate immune response (Fig. 21.2).
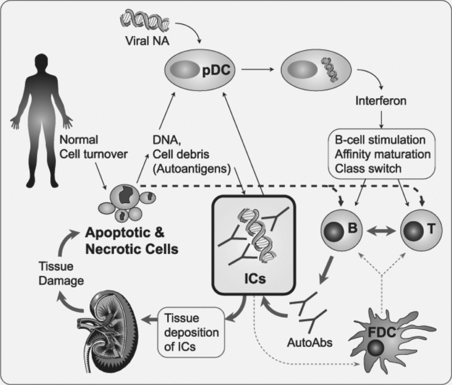
Dysfunctional overactivity of the type I interferon system is considered a central factor in SLE.11–13 The cell that is the most potent producer of type I interferons is the plasmacytoid dendritic cell (pDC), and the trigger for type I interferon production by pDCs is typically viral DNA and/or RNA in response to infection. Immune complexes composed of nucleic acids and IgG from SLE patients are potent inducers of type I interferon production by pDCs.14 The nucleic acids responsible for interferon production can be released from apoptotic or necrotic cells. Efficient production of type I interferons by SLE IgG requires Fcγ receptors and intact IgG, whereas F(ab) or F(ab′)2 fragments of IgG are not sufficient.15 Thus, a key mechanism leading to sustained production of interferon in SLE is thought to be immune complexes containing nucleic acids, including most prominently DNA and antiDNA and possibly RNA and antibodies to RNA-protein complexes. Internalization of the immune complexes is mediated by Fcγ receptors, and the internalized nucleic acid then binds to toll-like receptors (TLRs), including TLR 9. Binding of characteristic CpG motifs to TLR9 induces cellular signaling pathways leading to enhanced interferon mRNA transcription and protein release. ICs binding to germinal center follicular dendritic cells (FDCs) facilitates antigen presentation by FDCs and thereby promotes the ability of FDCs to interact with B-cells and cause affinity maturation and class switching. It has been proposed that immune-complex-bearing FDCs may be required for development of high-affinity IgG antibodies, including antiDNA.16 Thus, nucleic acid containing immune complexes augments the autoimmune response in SLE by leading to sustained overproduction of type I interferon and affinity maturation and continued production of antiDNA and related autoantibodies. Depressed clearance of immune complexes, as discussed in material following, may magnify this response by leading to higher levels of circulating immune complexes and/or higher binding of immune complexes to dendritic cells.
The type I interferon expression profile correlates with SLE disease activity, in general, with particularly close correlation with the presence and levels of autoantibodies to nucleic acids.17,18 These correlations have been used to support the notion that type I interferon overexpression leads to SLE. Conversely, this association also lends support to the hypothesis that immune complexes containing autoantibodies to nucleic acids lead to the SLE phenotype by causing enhanced type I interferon production. Whether the type I interferon profile is caused by antibodies to nucleic acids or vice versa, the association between the two types of abnormalities suggests an active role of both in the immune and inflammatory dysregulation found in SLE.
Immune Complex Clearance
The mononuclear phagocyte system plays the central role in removing immune complexes from the circulation, with clearance mediated by families of Fc and complement receptors on mononuclear phagocytes, neutrophils, and other cells. The presence of C3 receptors on primate erythrocytes but not erythrocytes from other species suggests a trafficking mechanism applicable to humans but not to non-primate experimental animals.19,20 Immune complexes that had activated complement and bound C3 in the circulation could bind to the complement receptor CR1 on the erythrocyte would be transported to the liver and spleen while bound to the red cell, and those immune complexes would be phagocytized by cells of the mononuclear phagocyte system (primarily via Fc receptors). In the liver, Kuppfer cells serve this phagocytic role. In the spleens of humans and some other species (but not mouse, rat, guinea pig, or rabbit), splenic filtering and immune complex trapping may be carried out at least in part in splenic ellipsoids, which are structures consisting of specialized capillary segments surrounded by macrophages.21
A variety of probes have been employed to determine the kinetics and sites of immune complex clearance experimentally in humans. Investigators have used erythrocytes coated with IgG antibodies, aggregated IgG, preformed immune complexes, and antigens infused into preimmunized subjects. Davies and colleagues have performed studies using several different soluble immune complexes as probes, including tetanus/antitetanus, hepatitis B surface antigen/antibodies, and murine IgG/human anti-mouse IgG.22 The former two types of immune complexes were formed in vitro and then injected into subjects. When soluble immune complexes of hepatitis surface antigen and antibody are made intentionally “small” (such as not to fix complement efficiently and which therefore do not bind to complement receptors on red cells), >90% are cleared by the liver [with a median clearance half-time of approximately 3 minutes (range 1 to 6 minutes)].23 The clearance half-time did not differ between normal individuals and subjects with SLE.
In other studies, complement depletion led to accelerated clearance of immune complexes by the liver and spleen and might have been associated with increased tissue deposition of immune complexes,24 which suggested to the authors that red cell binding of immune complexes could have role in “buffering” excessive loads of immune complexes until they are removed by mononuclear phagocytes. Others have suggested that erythrocyte binding of immune complexes could have a role in immune complex processing or degradation while on the erythrocyte.25 However, C1q-deficient mice also demonstrate an initial accelerated hepatic clearance of immune complexes and reduced splenic clearance.26 Because mice lack erythrocyte complement receptors, the accelerated hepatic uptake in C1q-deficient mice is not likely to depend on erythrocytes, indicating that complement modulates immune complex clearance by other mechanisms.
Davies and colleagues27 administered murine IgG and human antimouse IgG to study immune complexes formed in vivo, an experiment that might be considered most representative of natural physiology. Patients with ovarian carcinoma were given 131I-murine monoclonal antitumor antibodies and subsequently 125I-human antimouse IgG. Immune complexes were large but of a size possibly to be encountered physiologically. Soluble immune complexes formed within 5 minutes, activated complement, and were cleared with a half-life of 11 minutes in the liver and without a detectable increase in radioactivity over the spleen. Between 8 and 11% of the total available immune complexes bound to the erythrocyte, and at the time of peak red cell binding erythrocyte-bound immune complexes constituted approximately 20% of total circulating complexes. The majority of soluble immune complexes were cleared by mechanisms largely independent of red cells, and the site of clearance of these soluble complexes in the liver differed substantially from the splenic clearance of sensitized erythrocytes previously reported.28
In SLE patients, several studies have shown that the clearance of antibody-sensitized erythrocytes is slower than the clearance in normal controls, and slower in patients with active renal disease than in those without.29,30 Investigators at Leiden have administered radioiodinated aggregated human IgG (123I-AHG) to SLE patients to explore the fate of circulating soluble immune complexes in patients with SLE. The investigators described an initial rapid clearance and later slower clearance of immune complexes from the circulation (both reported in terms of the time to removal of 50% of the maximum material, T1/2). In their first study, the authors reported that the initial phase T1/2 was not significantly different between SLE patients and controls, whereas the second phase T1/2 was prolonged in the patient group.31
In the second study, SLE patients erythrocytes were observed to have a decreased number of CR1, which was associated with less binding of AHG to red blood cells and with a faster initial rate of clearance of AHG (mean half-time to removal 5.2 ±0.2 minutes in patients versus 6.6 ±0.2 minutes in controls, p = 0.01). The later phase of AHG clearance was similar in patients and controls (T1/2 148 ±18 versus 154 ±20 minutes). Both the maximum liver uptake and time required to reach the maximum liver uptake were similar in SLE patients and controls. Of interest, the feature most predictive of the rate of AHG clearance in SLE patients was the serum IgG concentration, which was inversely correlated (r = −0.66) with the rate of clearance. The authors speculated that the concentration of serum IgG in SLE patients was a primary determinant of the proportion of Fc receptors occupied, and thereby governed the rate of clearance of AHG.32
The importance of the rapid, very early removal of immune complexes from the circulation was shown by Schifferli and colleagues, who examined the clearance of immune complexes composed of tetanus toxin and anti-tetanus in 4 patients with SLE, as well as 11 other patients and 9 normal subjects.33 The authors reported that the removal of these large complexes from the circulation occurred in two phases: a very rapid “trapping” phase that occurred within the first minute and a monoexponential later phase. In 1 of 9 normal individuals and 11 of 15 patients, over 8% of the injected immune complexes were removed from the circulation (“trapped”) within the first minute after administration, a time point and amount removed that could not be attributed to clearance by the liver and spleen and therefore trapping presumably resulted in deposition of immune complexes in peripheral tissues. This initial trapping was seen in patients with serum complement deficiencies, and was associated with lower levels of CR1 on erythrocytes. The later phase of immune complex clearance was exponential over the 60 minutes of measurement, with between 9.9 and 18.7% removed per minute in normals and 8.6 to 32.2% per minute removed in patients. When opsonized immune complexes bound in vitro to erythrocytes via CR1 were injected into patients there was release of 10 to 81% of the immune complexes from the erythrocytes within 1 minute of injection. The extent of this release was inversely correlated with CR1 number/cell.
Together, these studies of clearance of soluble immune complexes in SLE patients argue that the hepatic clearance of immune complexes (which governs the late-phase removal of soluble immune complexes) is probably normal in SLE patients. Low CR1 numbers on erythrocytes or profound hypocomplementemia can permit deposition of immune complexes within tissues during the early phase of immune complex clearance. Reduction in CR1 numbers is an acquired abnormality associated with active SLE.34 It is unclear the degree to which the abnormalities in immune complex clearance mechanisms observed in these experiments contributes to immune complex deposition at sites of tissue injury.
More recently, investigations have explored the implications of polymorphisms in various Fcγ receptors with regard to their potential role in clearing immune complexes from the circulation and causing a predisposition to SLE. Lack of the H131 allele of the FcγRIIA, which is responsible for efficient clearance of IgG2-containing immune complexes, has been associated with lupus nephritis in American blacks.35 A report has implicated a functionally important genetic polymorphism of FcγRIIIA as a risk factor for SLE in a genetically diverse group of patients.36
Factors Governing Immune Complex Localization: Physicochemical Composition and Site of Formation
In experimental models, administration of preformed immune complexes results in mesangial and subendothelial localization of immune complexes within renal glomeruli. Studies in the 1980s using the Heymann model of nephritis and studies on isolated perfused kidneys emphasized that antibodies and antigens could deposit sequentially in the kidney, with the result that the immune complexes form in situ and tend to localize in the subepithelial region of glomeruli rather than being deposited from circulation.37 Formation of complexes in situ can occur because of direct binding of antigens or antibodies, initially because of interaction between the circulating molecule and structures within the kidney. This initial interaction can be relatively weak and/or nonspecific (e.g., because of charge-charge interactions).
Electrical charge on either the antigen or the antibody within the immune complex governs interaction with fixed negative charges on proteoglycans in the basement membrane or in other structures and influences both the deposition and persistence of antigens, antibodies, and immune complexes in tissues. In experimental systems, even a small proportion of positively charged (cationic) antibodies enhance binding and persistence of immune complexes in renal glomeruli.38
Deposition of antigens or antibodies could be augmented or facilitated also by antigen-specific receptors within the tissues. Particularly relevant for the study of SLE, Emlen and Burdick demonstrated that immune complexes containing DNA may be removed in part by DNA receptors.39 In experimental animals, the clearance of immune complexes containing glycosylated antigens is governed in part by specific carbohydrate receptors on hepatocytes.40 A serum carbohydrate binding protein, mannose-binding protein (MBP), may have an important role in clearing immune complexes containing antigens with selected carbohydrate residues. A member of the collagen motif-containing collectin family of proteins, MBP binds terminal mannose, fucose, glucose, fucose, or N-acetylglucosamine residues; can activate the classical or alternative pathways of complement;41 can activate macrophages via the C1q receptor;42 and can serve as an opsonin.43
Genetic polymorphisms responsible for depressed function and serum levels of MBP are associated with SLE in African Americans44 and other groups.45,46 Furthermore, certain ribonucleoprotein autoantigens (including the U1-specific 68kD and A proteins and the U2-specific B′ protein) are glycoproteins, with mannose, glucose, and N-acetylglucosamine detected on the 68kD protein.47 Thus, it is conceivable that the clearance of glycoprotein antigens or immune complexes containing such antigens (including the U1-RNP particle) could be influenced by MBP polymorphisms. These considerations suggest that MBP polymorphisms could participate in the pathogenesis of SLE by influencing immune complex clearance, analogous to the role of polymorphisms in complement components and FcR.
Features on the antibodies within the immune complex can influence the physiology of immune complexes. The isotype of antibodies influences immune complex handling, in that the ability to activate complement influences both the ability to bind to complement receptors as well as to activate inflammatory cascades. AntidsDNA in SLE patients tend to be of subclasses IgG1, IgG2, and IgG3, and tend to be efficient in activation of complement.48 Experimental studies with murine monoclonal IgG3 immunoglobulins have demonstrated that deposition of cryoprecipitating or other self-associating immunoglobulin aggregates, a feature of certain immunoglobulin molecules, may cause glomerulonephritis.49 IgA-containing immune complexes may be cleared by distinct IgA receptors.50
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


