Chapter 9 Hormonal Influences in the Expression of Systemic Lupus Erythematosus
INTRODUCTION
The ability of the immune system to distinguish self from non-self is essential to maintain nonresponsiveness to self. Although self-recognition occurs during development, checkpoints that operate in the adult are critical for maintaining and establishing tolerance to self-antigens that appear after maturity.1 The process of self-tolerance continues in adult life. In autoimmune diseases, it is postulated that self-reactive cells expressing antigen receptors escape clonal deletion.2 Moreover, somatic mutations can potentially generate autoreactive antigen receptors.3 Loss of self-recognition, or autoimmunity, is more prevalent in females than in males.4 The timing of disease onset for some autoimmune disorders, such as lupus, occurs after puberty—suggesting that sex steroids could influence the gender bias of disease onset.5 The correlation between sexual maturity and some autoimmune diseases is consistent with the view that sex steroids enhance or suppress molecules that regulate pathways important in maintaining tolerance to self-antigens. In general, estrogens have been shown to promote or stimulate the immune response, whereas androgens and progesterone are considered more suppressive6–8 (Table 9.1).
TABLE 9.1 HORMONES AND SOME OF THEIR EFFECTS ON THE IMMUNE SYSTEM
| Hormone | Role in Immune System | Gene Targets |
|---|---|---|
| Estrogen (estradiol 17-β) | Immunostimulatory, protective for some autoimmune diseases, activates B-& T-cells, decrease/increase apoptosis | |
| Progesterone | Immunoprotective, gamma/delta TCR+ | |
| Androgens | Immunoprotective, B and T lymphocytes, thymic atrophy, thymocyte apoptosis | TGF-β, macrophage migration inhibitory factor |
| Dehydroepiandrosterone (DHEA) | Stabilizes disease symptoms in SLE, androgen precursor, anabolic steroid | |
| Prolactin | Stimulates disease activity in SLE, T-cell proliferation, B-cell maturation | Interferon regulatory factor-1, Bcl-2, CD40 |
Hormones alter the expression of a variety of genes that control the proliferation, differentiation, and apoptosis of lymphocytes, macrophages, and dendritic cells of the immune system. Hormones also regulate the production of cytokines and cell adhesion molecules. Changes in the expression of many of these gene targets have been associated with pathogenesis in autoimmune diseases but the mechanisms involved require clarification.
SEXUAL DIMORPHISM AND AUTOIMMUNITY
Several autoimmune diseases (including SLE, Sjögren syndrome, multiple sclerosis, rheumatoid arthritis, Graves’ disease, and Hashimoto thyroiditis) are sexually dimorphic because they occur more frequently in women than in men.4 In murine models of autoimmune diseases such as autoimmune encephalomyelitis and collagen-induced arthritis, females typically respond more robustly than males. The factors contributing to the development of autoimmune disease appear to involve lymphocyte-mediated mechanisms that control disease pathogenesis, activation, or regression. Although it is still not known how sex hormones influence cells in the immune system, emerging evidence suggests that hormones affect cellular differentiation, cytokine production, T helper (Th)1 and Th2 cell polarization, MHC class 2 expression, and antigen-presenting cell recruitment or function8–10 (Table 9.1).
Females have more immunoglobulins in circulation than males. Estrogen increases in vitro production of IgG and IgM in isolated human peripheral blood mononuclear cells.9 Estrogen may also affect antigen presentation by stimulating the differentiation of dendritic cells and the proliferation of naive CD4+ cells.11 Dendritic cells from mice induced to develop experimental allergic encephalomyelitis show a reduced capacity to present specific antigen to T-cells when cultured in medium containing estradiol. These cells express less TNF-a, interferon gamma, and IL-12 (suggesting that estrogen stimulates a shift to the Th2 phenotype and reduces inflammation).12 We have shown that estrogen suppresses TNF-a production by lupus T-cells in vitro and that it reduces apoptosis.13
HORMONES AND THE IMMUNE SYSTEM
Hormones are chemical messengers secreted into the blood stream that bind to specific receptor proteins in the nucleus (steroid hormones) or on the surface (peptide hormones) of target cells. Prolactin is a hormone that is synthesized and secreted from the anterior pituitary gland. Receptors for prolactin are expressed on T- and B-cells,14 and 20 to 30% of patients with SLE have increased levels of prolactin in circulation.15,16 Treatment of NZB/NZW F1 mice with the prolactin inhibitor bromocriptine improves survival of the mice,17 suggesting that prolactin stimulates disease progression in SLE. Prolactin exerts its effects, at least in part, on B-cells by rescuing autoreactive B-cells that would normally be targeted for apoptosis.18 Continuous administration of prolactin to mice transgenic for anti-DNA antibody heavy chain (R4A-IgG2b, BALB/c) over a period of four weeks led to a lupus-like phenotype.18 In these mice, prolactin increased B-cell survival, presumably by up-regulating Bcl-2 and protecting autoreactive cells from apoptosis.
Interestingly, prolactin effects are influenced by the genotype of these mice because R4A-IgG2b C57BL/6 mice maintain tolerance with the same prolactin treatment that induced a lupus phenotype in the R4A-IgG2b BALB/c mice. Prolactin levels in human females with SLE correlate with disease activity,16,19 and conventional treatment for SLE reduces disease activity and prolactin levels in these patients. Prolactin expression increases in response to estrogen, and the use of estrogen receptor (ER) selective agonists indicates that both ER-a and ER-β stimulate prolactin secretion in the rat.20 It is now important to test if hyperprolactinemia results from a defect in estrogen-dependent regulation of the prolactin gene or if increased secretion of prolactin in SLE patients is controlled by other factors.
Dehydroepiandrosterone (DHEA) and its sulfate (DHEAS) are the main adrenal androgens produced in humans.21 DHEA is unique compared with other adrenal steroids because the levels in circulation fluctuate from birth into advancing age.22 DHEA is a precursor for sex hormone synthesis and acts as an anabolic steroid.23 Pharmacologic studies suggest that DHEA therapy increases testosterone levels in women but not in men.22 SLE patients treated with prednisone have low levels of DHEA.23 In SLE patients with mild disease, DHEA improves or stabilizes disease symptoms.24,25
ESTROGENS AND THE IMMUNE SYSTEM
The action of estradiol in the classical target tissues of the uterus and the breast has been extensively studied.26–28 In classical target cells, estradiol increases protein synthesis, stimulates proliferation, induces the production of growth factors and their receptors, and recruits infiltration of macrophages into target tissues such as the uterus. We are just beginning to understand how estradiol action can stimulate the immune system.29–31 It seems likely that estradiol will affect processes in target cells of the immune system in a manner similar to classical endocrine cell types, although the target genes may be different. Recent studies of endocrine effects on cells of the immune system support the growth-promoting effects of estradiol through increased production of cytokines32,33 and immunoglobulins in circulation.34,35
Estradiol enhances the proliferation of T-cells36 and macrophages37 and affects apoptosis of autoreactive B-cells.29,31,38 There is also exciting new evidence that suggests activation of the ER can be compartmentalized to specific cell types of the immune system.39–41 Mice lacking functional ER-a by target mutagenesis were induced to develop experimental allergic encephalomyelitis.39 Inflammation of the central nervous system (CNS) was initiated by autoantigen-specific T lymphocytes located in the CNS compartment of these mice. The authors39 suggest that estrogen may play a role in activating the vascular endothelium of the blood/brain barrier and inhibit T-cell adhesion in the CNS. Taken together, the emerging evidence suggests that estrogen regulates genes that promote growth and activity of cells that function in the immune system. Because estrogen is a key regulator of molecules involved in inflammation, any organ system in which inflammation pathways are activated is a likely target for estrogen action.10,42,43
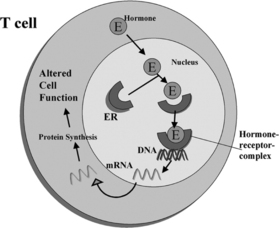
Estrogen enters target cells from the circulation, and once in the cell the hormone binds to high-affinity receptor proteins (Fig. 9.1). The estrogen-dependent changes in cell behavior occur in response to the binding of the ligand-receptor complex to specific DNA sites that induce or suppress gene transcription.26,44–46 At least some of the potential gene targets in cells comprising the immune system have been identified.29,30,47 Estrogen up-regulates the expression of genes that are markers for B-cell and T-cell activation. Estrogen directly stimulates the expression of genes in mouse B-cells, which allows some autoreactive cells to escape B-cell-mediated apoptosis.29 Estrogen action thus rescues a population of autoreactive B-cells that would be deleted from the repertoire in the absence of the hormone. We have shown that two markers of T-cell activation, calcineurin and CD154, are up-regulated by estrogen in SLE but not in normal T-cells.48,49 Abnormal T-cell regulation of B-cells results in antibody/autoantibody secretion.50,51 SLE is a disorder that results from a loss of self-recognition, and the resulting secretion of autoantibodies leads to a variety of pathologic outcomes.52,53
Because estrogen can both stimulate and inhibit gene expression, it is not surprising that for some autoimmune diseases estrogen may be protective. The clinical assessment of various diseases suggests that estrogen can be beneficial in autoimmune disorders, including multiple sclerosis, rheumatoid arthritis, uveitis, and thyroiditis. These autoimmune diseases improve during pregnancy most likely owing to estrogen influence on inducing a shift from Th1 to Th2 cytokine profiles during gestation.54 In experimental autoimmune encephalomyelitis (EAE), a mouse model for human multiple sclerosis, estrogen decreased production of proinflammatory cytokines (including tumor necrosis factor, interferon gamma, and IL-2).55 The protective effect in these autoimmune mice was postulated to be due to down-regulation of proinflammatory cytokines and an estrogen-dependent increase in Th2 cytokines such as IL-5.55 Mice induced to develop EAE benefit by the administration of low levels of estrogen prior to induction of the disease.56
One of the mechanisms involved in this protective effect of estrogen pretreatment is postulated to be an increase in the expression of Foxp3 and the subsequent regulation of genes involved in the maintenance of tolerance to self-antigens.40,56 Foxp3 appears to be a master controller of regulatory T-cells because increased expression of the FOXP3 protein stimulates expansion of CD4+ CD25+ T-cells.40 This regulatory T-cell subset is thought to play an essential role in the maintenance of tolerance.40,57 Several observations suggest that FOXP3 may exert its effects by transcriptional repression because it decreases the expression of IL-2 and may down-regulate a variety of other cytokines, including IL-4, TNF, and granulocyte macrophage colony-stimulating factor.57 Additional studies are now warranted to investigate the estrogen-dependent regulation of Foxp3 in gender-biased autoimmune diseases such as lupus.
ESTROGEN RECEPTORS
Sex steroid receptors are expressed in most cells that comprise the immune system.30,58–60 However, their function in the immune system remains to be clearly established. Steroid receptors are part of the nuclear receptor gene family that contains a large group of homologous proteins, including the steroids, thyroid hormone, retinoic acid, and vitamin D.44–46 In the general scheme of nuclear hormone receptor action, the hormone or ligand binds to the receptor and this interaction stimulates a conformational change that leads to DNA binding at specific sequences along target genes (see Fig. 9.1). Evidence suggests that the interaction of activated receptors with target genes can both increase and decrease transcription and thereby alter cell function.
Although this classical mechanism of sex steroid action is still considered a valid model, more recent evidence indicates that additional layers of complexity are required for appropriate signaling events activated by nuclear receptors. It is now evident that there are a host of cofactors that interact with the receptors and modulate gene transcription.61 Although this is a relatively unexplored area of research in cells comprising the immune system, it is logical to assume that cofactors will be major modulators of hormone-dependent gene regulation in all cells that express steroid receptors. In this chapter, we focus specifically on information relating to the steroid hormone estrogen and the ER because compelling evidence is emerging for estrogen effects on the development and pathogenesis of autoimmune diseases.30,31,62–64
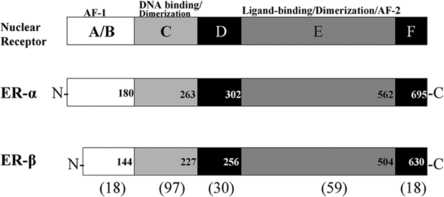
Estrogen effects are exerted through two receptor proteins termed estrogen receptor-alpha (ER-α) and estrogen receptor-beta (ER-β). The receptors are coded for by separate genes, but the proteins exhibit regions of high homology in the DNA and ligand binding domains.27,28 ER subtypes are modular in structure, with functional domains characteristic of all nuclear receptors (see Fig. 9.2). The amino termini of the ERs have diverged and only share about 18% homology. This divergent region may be important for the differential action of ER-a and ER-β in cells that coexpress both receptors.65,66 There is evidence that the magnitude of an estrogenic response to ER-a and ER-β activation varies depending on promoter context and cell type.66 Evidence suggests that ER-a and ER-β function is not redundant in cells in which receptor subytpes are coexpressed. It is now important to investigate how differential receptor subtype action influences target cell behavior and if altered function underlies the development of endocrine-dependent autoimmune disease.
< div class='tao-gold-member'>
Related posts:
 Assessment of Disease Activity in Systemic Lupus Erythematosus
Assessment of Disease Activity in Systemic Lupus Erythematosus
 The Environment in the Pathogenesis of Systemic Lupus Erythematosus
The Environment in the Pathogenesis of Systemic Lupus Erythematosus
 Antibodies and their Antigenic Targets in the Antiphospholipid Syndrome
Antibodies and their Antigenic Targets in the Antiphospholipid Syndrome
 Complement Deficiencies in Human Systemic Lupus Erythematosus (SLE) and SLE Nephritis: Epidemiology and Pathogenesis
Complement Deficiencies in Human Systemic Lupus Erythematosus (SLE) and SLE Nephritis: Epidemiology and Pathogenesis
 What Do Mouse Models Teach Us about Human Systemic Lupus Erythematosus?
What Do Mouse Models Teach Us about Human Systemic Lupus Erythematosus?
 Infection and Autoimmunity
Infection and Autoimmunity
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


