Chapter 54 Homocysteine Metabolism
Nutritional Modulation and Impact on Health and Disease
 Introduction
Introduction Homocysteine Metabolism
Homocysteine Metabolism
Gender and Genetics
Studies of healthy men and women indicate that certain acquired and genetic determinants may affect total plasma homocysteine. Women tend to have lower basal levels than men,1 and neither contraceptives nor hormone replacement therapy seem to alter their levels significantly.2 However, in postmenopausal women, hormone replacement therapy might slightly decrease elevated homocysteine concentrations. No significant lowering effect was observed in women with low homocysteine levels.3 Generally, homocysteine concentrations are significantly higher in postmenopausal women than in premenopausal women; however, the previously mentioned sex differences in homocysteine concentrations persist even in elderly populations.4–6 The antiestrogen drug tamoxifen, used in the long-term treatment of breast cancer patients, was reported to decrease homocysteine levels in postmenopausal women with breast cancer.7
A number of reports also demonstrated that elevated plasma total homocysteine in children was correlated to either cardiovascular disease or death in their parents or close relatives. This relationship was observed in both white and black children, and in white children with hypercholesterolemia. In the latter study group, the 5,10-methylenetetrahydrofolate reductase (MTHFR) TT genotype tended to be most frequent in children with a parental history of cardiac disease.8 Epidemiologic evidence showed homocysteine levels to be more than 45% lower in Westernized adult black Africans than in age-matched white adults, revealing racial genetic differences in homocysteine metabolism.9
The MTHFR gene has two different alleles where the “T” allele is associated with decreased enzyme activity, hyperhomocysteinemia, and increased risk for thromboembolism in coronary heart disease. The presence of the “C” allele is correlated to lower homocysteine levels and, not surprisingly, may even provide protection against occlusion of coronary arteries. One Hungarian study concluded that the carriers of the T allele with coronary heart disease died earlier due to myocardial infarction.10
Lifestyle
An association between coffee and black tea consumption and the concentration of total homocysteine in plasma has been reported.11 A marked positive dose–response relation between coffee consumption and plasma homocysteine levels was observed. The relationship was most marked in males and females consuming more than eight cups of coffee per day. The combination of cigarette smoking and high coffee intake was associated with particularly high homocysteine concentrations.12 Long-term ingestion of alcohol has also been associated with increased homocysteine levels.13,14 Plasma total homocysteine is inversely related to exercise.15
Nutritional Relationships
Nutrition affects homocysteine concentrations in both men and women. For example, individuals in the lowest quartiles for serum folate and vitamin B12 have significantly higher concentrations of homocysteine, and men in the lowest quartile of serum pyridoxal 5´-phosphate (P5P; the bioactive form of vitamin B6) also have increased homocysteine concentrations.2
Methionine
Homocysteine is an intermediate product of methionine metabolism and is metabolized by two pathways: the remethylation pathway, which regenerates methionine, and the transsulfuration pathway, which degrades homocysteine into cysteine and then taurine. In essence, the intermediate metabolite homocysteine is located at a metabolic crossroads, so it directly and indirectly affects all methyl and sulfur group metabolism occurring in the body. Experiments demonstrated that high levels of L-homocysteine and adenosine in the cell inhibit all methylation reactions.16
The remethylation pathway (Figure 54-1) comprises two intersecting biochemical pathways and results in the transfer of a methyl group (CH3) to homocysteine by either methylcobalamin or betaine (trimethylglycine). Methylcobalamin originally receives its methyl group from S-adenosylmethionine (SAMe) or 5-methyltetrahydrofolate (5-methylTHF), an active form of folic acid. After remethylation, methionine can be reused to produce SAMe, the body’s “universal methyl donor,” which participates in several key metabolic pathways, including methylation of DNA and myelin, synthesis of carnitine, coenzyme Q10 (CoQ10), creatine, epinephrine, melatonin, methylcobalamin, and phosphatidylcholine (PC), as well as phase 2 methylation detoxification reactions.
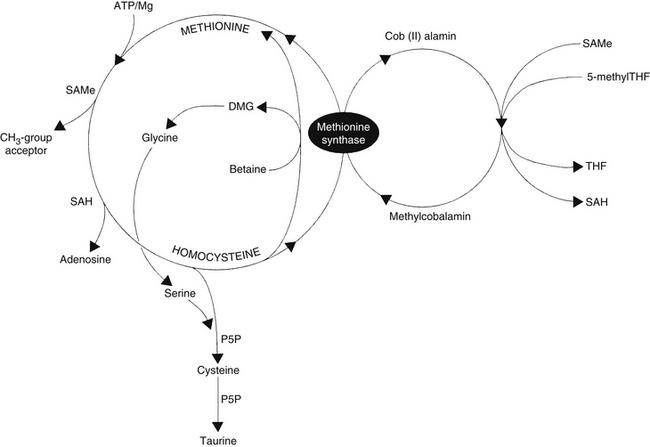
The transsulfuration pathway of methionine/homocysteine degradation (see Figure 54-1) produces the amino acids cysteine and taurine, which are important nutrients for cardiac health, hepatic detoxification, cholesterol excretion, bile salts formation, and glutathione (GSH) production. This pathway depends on adequate dietary intake and hepatic conversion of vitamin B6 into its active form, P5P. The amino acid serine, a down-line metabolite generated from betaine via the homocysteine remethylation pathway, is also necessary.
In addition to 5-methylTHF, methylcobalamin, betaine, and P5P, N-acetylcysteine (NAC) has been reported to significantly lower homocysteine levels.17
S-Adenosylhomocysteine
The metabolic precursor of homocysteine in all tissues is S-adenosylhomocysteine, which is more difficult to measure than homocysteine, but has recently been indicated as a more sensitive marker for cardiovascular disease and Alzheimer’s disease. S-adenosylhomocysteine is formed by the demethylation of SAMe (Figure 54-2). S-adenosylhomocysteine hydrolase catalyzes the hydrolysis of S-adenosylhomocysteine to adenosine and homocysteine. S-adenosylhomocysteine hydrolase deficiency is a genetic disorder of methionine metabolism that results in slow psychomotor development.
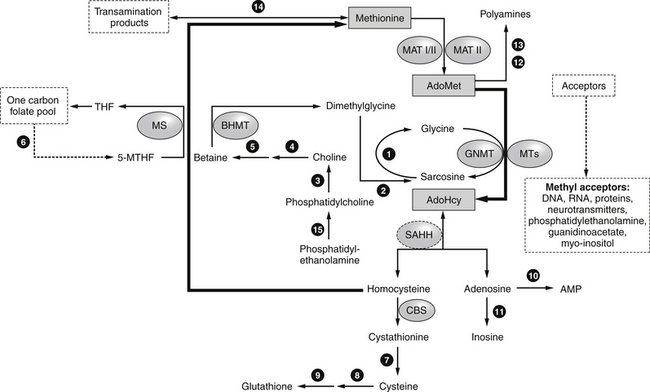
FIGURE 54-2 Methionine, AdoMet (S-adenosylmethionine), and AdoHcy metabolism.
(From Baric I, Fumić K, Glenn B, et al. S-adenosylhomocysteine hydrolase deficiency in a human: a genetic disorder of methionine metabolism. Proc Natl Acad Sci, 2004:101:4234-4239.)
Increased levels of S-adenosylhomocysteine may occur when the intracellular concentration of homocysteine increases.18–20
Methyltetrahydrofolate
Synthesis of the active forms of folic acid is a complex process requiring several enzymes, as well as adequate supplies of niacin, P5P, and serine as cofactors (Figure 54-3). In plants, folic acid is formed from a hetero-bicyclic pteridine ring, para-aminobenzoic acid, and glutamic acid. Folate is initially deconjugated in the cells of the intestinal wall to the monoglutamate form. This is then reduced to dihydrofolate and then to tetrahydrofolate (THF) via folate and dihydrofolate reductase. Both enzymes require reduced nicotinamide adenine dinucleotide phosphate (niacin dependent) as a cofactor. Serine combines with P5P to transfer a hydroxymethyl group to THF. This results in the formation of 5,10-methylenetetrahydrofolate (5,10-methyleneTHF) and glycine. This molecule is of central importance, being the precursor of the metabolically active 5-methylTHF, which is involved in homocysteine metabolism, and 10-formyltetrahydrofolate (involved in purine synthesis), as well as functioning on its own in the generation of thymine side chains for incorporation into DNA.
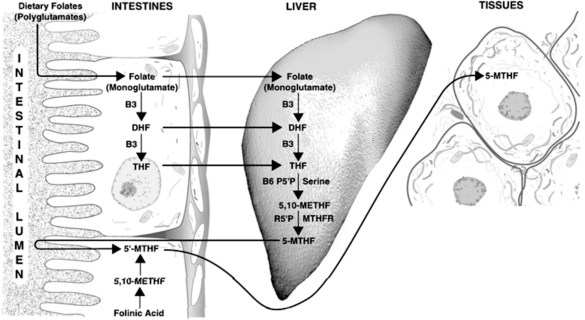
The following may contribute to a deficiency of folic acid:
• A defect in utilization, as in alcoholics
• Increased needs in pregnant women and cancer patients
• Metabolic interference by drugs
• Folate losses in hemodialysis
• Enzyme or cofactor deficiency necessary for generation of active folic acid
Individuals using supplements or consuming either breakfast cereals or green leafy vegetables have significantly greater plasma folate and lower homocysteine levels than those who do not.1
Folinic acid (5-formylTHF), available supplementally as calcium folinate (also known as leucovorin calcium), is an immediate precursor to 5,10-methyleneTHF and 5-methylTHF. Folinic acid can correct deficiencies of the active forms of folic acid, is more stable than folic acid, and has a longer half-life in the body. Folinic acid also readily crosses the blood–brain barrier and is slowly cleared, compared with folic acid, which is poorly transported into the brain and, once in the central nervous system (CNS), is rapidly cleared.21
Methylcobalamin
Methylcobalamin’s only known biological function in humans is in the remethylation of homocysteine to methionine via the enzyme methionine synthase, also known as 5-methyltetrahydrofolate-homocysteine methyltransferase. To originally form methylcobalamin from cyanocobalamin or other Cob(III)alamin or Cob(II)alamin precursors, SAMe must be available to supply a methyl group. Once methylcobalamin is formed, it functions in the regeneration of methionine by transferring its methyl group to homocysteine. Methyl-cobalamin can then be regenerated by 5-methylTHF (Figure 54-4). The cell’s ability to methylate important compounds such as proteins, lipids, and myelin is compromised by a deficiency of either folate or vitamin B12.22 Shortages of active folic acid, SAMe, or a dietary deficiency of cobalamin lead to a decrease in the generation of methylcobalamin and a subsequent impairment of homocysteine metabolism. Because lack of methylcobalamin leads to depressed DNA synthesis, rapidly dividing cells in the brain and elsewhere are affected.
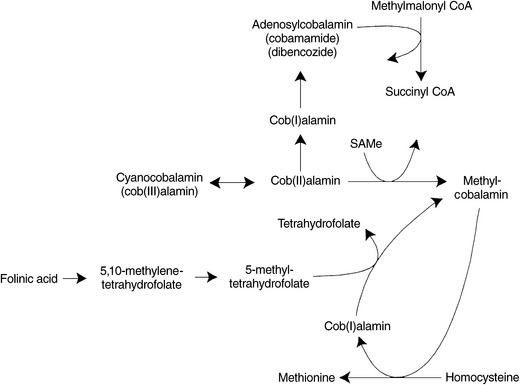
At least 12 different inherited inborn errors of metabolism related to cobalamin are known. Abnormalities are detectable by urine and plasma assays of methylmalonic acid and homocysteine; and plasma and erythrocyte analysis of cobalamin coenzymes, which can reveal deficiencies of methylcobalamin or adenosylcobalamin.23
Betaine
Betaine donates one of its three methyl groups via the enzyme betaine-homocysteine methyltransferase to homocysteine, resulting in the regeneration of methionine. After the donation of the methyl group, one molecule of dimethylglycine remains. This molecule is oxidized to glycine and to two molecules of formaldehyde by riboflavin-dependent enzymes. The formaldehyde can combine with THF within the mitochondria to generate one of the active forms of folic acid, methylenetetrahydrofolate, which can be converted to 5-methylTHF and subsequently used as a methyl donor (Figure 54-5).
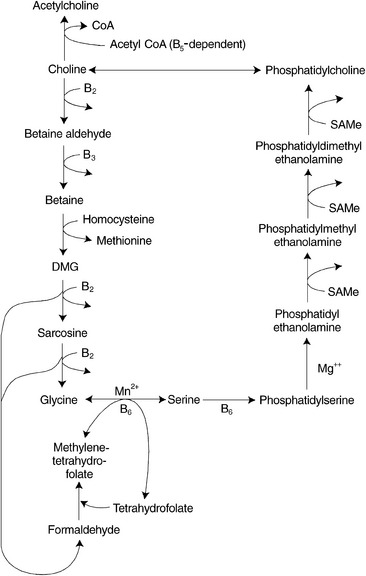
In animal studies, a disturbance in the metabolism of either of the two methyl-donor pathways, due to limited availability of either betaine or folates and vitamin B12, affects levels of nutrients in the coexisting pathway, because more of a drain is placed on the other pathway as a source of methyl groups. Rats fed diets deficient in choline and methionine had hepatic folate concentrations half that of controls after 5 weeks.24 During choline deficiency, hepatic SAMe concentrations were also shown to decrease by as much as 50%.25 Similarly, THF deficiency resulted in decreased hepatic total choline levels.26
Patients with a congenital deficiency of the enzyme MTHFR, which is needed for the formation of 5-methylTHF, have reduced levels of both methionine and SAMe in the cerebrospinal fluid and show demyelination in the brain and degeneration of the spinal cord. Methionine is effective in the treatment of some of these patients; however, betaine was shown to restore cerebrospinal fluid SAMe levels to normal and to prevent the progress of neurologic symptoms in all patients in whom it was tried.27
Betaine supplementation has been shown to reduce homocysteine levels28 while resulting in modest increases of plasma serine and cysteine levels.29 Stimulation of betaine-dependent homocysteine remethylation causes a commensurate decrease in plasma homocysteine that can be maintained as long as supplemental betaine is taken.30
Serine levels are depressed in some individuals with excess homocysteine who are treated with folic acid, cobalamin, and vitamin B6.31 Because serine is required (1) for the conversion of folic acid to its active form, (2) as a shuttle for methyl groups between the cytosol and the mitochondria, and (3) as a cofactor in the transsulfuration pathway of methionine/homocysteine metabolism, supplementation with betaine should be included with folic acid, cobalamin, and P5P to optimize the interrelated pathways of homocysteine metabolism.
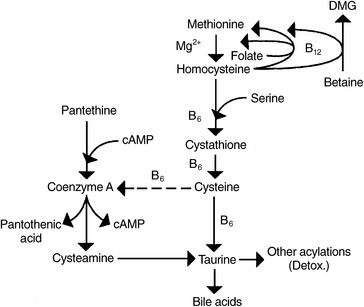
Pyridoxal 5´-Phosphate
P5P is the active coenzyme form of vitamin B6. This cofactor is involved in myriad biological processes, including the transsulfuration pathway of homocysteine. This degradation pathway involves a two-step process, resulting in the formation of cystathionine and its subsequent cleavage to cysteine. Cystathionine synthase and cystathioninase require P5P as a cofactor, and the committed first step in the degradation of homocysteine, cystathionine synthase, also requires serine, a downstream metabolite of betaine (Figure 54-6). Some studies suggest that cystathionine could be a useful marker to assess the effect of vitamin B6 and should be monitored with homocysteine to better elucidate clinical outcomes.32
 Renal Function
Renal Function
Impaired kidney function appears to be a factor in raised homocysteine concentrations.33 It has been shown that a decreased glomerular filtration rate may be a contributing factor, and some researchers suggest that hyperhomocysteinemia may actually reflect early nephrosclerosis,8 although other studies demonstrate only a minor renal influence.34 Fenofibrate treatment, known to increase homocysteine levels, is also known to promote elevations in serum creatinine (see “Pharmaceutic Drug Effects on Homocysteine”).35 Cystatin C, a creatinine-independent indicator of glomerular filtration rate, has also been shown to be well associated with homocysteine levels in kidney transplantation patients, as well as those with coronary disease.35,36
 Pharmaceutic Drug Effects on Homocysteine
Pharmaceutic Drug Effects on Homocysteine
Fibrates such as gemfibrozil are a class of drugs used to lower triglycerides and raise high-density lipoprotein levels. In a paradoxic effect working against a safer cardiac risk profile, these drugs have been observed to raise homocysteine levels up to 40%.35,37,38 The mechanism of this observation is unknown, although it seems that renal mechanisms may be involved (see “Renal Function”).
Thiazides and angiotensin-converting enzyme inhibitors are often a first-line conventional treatment to lower elevated blood pressure. One preliminary, randomized, prospective treatment study investigated 40 hypertensive patients after treatment with hydrochlorothiazide or captopril. In this study vitamins B6, B12, folic acid, creatinine, and cystatin C were used as parameters of renal function. For 31 and 29 days, respectively, 21 patients were prescribed hydrochlorothiazide and 19 were prescribed captopril. It was found that hydrochlorothiazide, but not captopril, significantly raised homocysteine by 16%, as well creatinine and cystatin C.39
 Impact of Homocysteine on Key Nutrients
Impact of Homocysteine on Key Nutrients
Carnitine
A trimethylated amino acid roughly similar in structure to choline, carnitine is a cofactor for transformation of free long-chain fatty acids into acyl-carnitines and their transport into a mitochondrial matrix, where they undergo β-oxidation for cellular energy production. Synthesis of carnitine begins with the methylation of the amino acid L-lysine by SAMe. Methionine, magnesium, vitamin C, iron, P5P, and niacin, along with the cofactors responsible for regenerating SAMe from homocysteine (5-methylTHF, methylcobalamin, and betaine), are required for optimal carnitine synthesis (Figure 54-7).
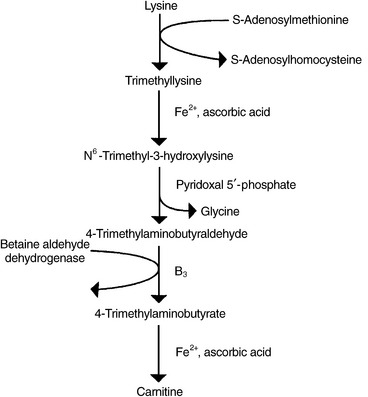
A pivotal enzyme in carnitine synthesis, betaine aldehyde dehydrogenase, is the same enzyme responsible for synthesis of betaine from choline. Studies suggest this enzyme has a preference for the choline–betaine conversion, and that choline supplementation may decrease carnitine synthesis; therefore, it may be of greater benefit to supplement with betaine rather than its precursor, choline.40,41
Chondroitin Sulfates, Glucosamine Sulfate, and Other Sulfated Proteoglycans
Proteoglycans are amino sugars found in all tissues, the highest being in cartilage, tendons, ligaments, synovial fluid, skin, fingers, toenails, heart valves, and the basement membrane of all blood vessels. Perhaps the most widely known of the amino sugars are the chondroitin sulfates and glucosamine sulfate (see Chapter 94 for further discussion).
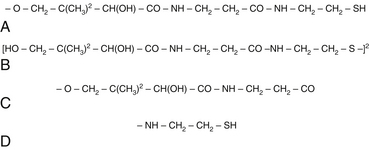
Coenzyme A
CoA consists of an adenine nucleotide and phosphopantetheine. Contained within the structure of this coenzyme is pantothenic acid; however, the reactive component of the molecule is a sulfhydryl group that is not contained within the vitamin. To form the sulfhydryl-containing molecule (pantotheine), pantothenic acid must combine with cysteamine. Cysteamine is formed through conjugation and decarboxylation reactions of cysteine. The disulfate form of pantetheine, known as pantethine, as opposed to pantothenic acid, bypasses cysteine conjugation and decarboxylation. This might account for some of the clinical benefits seen with pantethine supplementation that have not been reproduced with the supplementation of pantothenic acid (Figure 54-8, A to D, for the chemical structures of pantotheine, pantothine, pantothenic acid, and cysteamine).
Coenzyme Q10
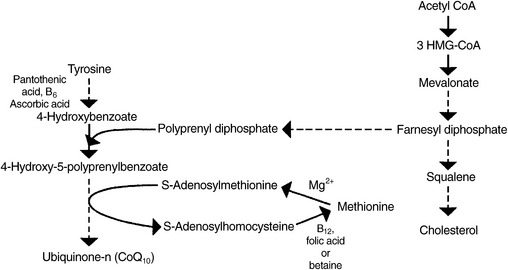
CoQ10 is a fat-soluble quinone occurring in the mitochondria of each cell (see Chapter 79 for a full discussion). The primary biochemical action of CoQ10 is as a cofactor in the electron transport chain, the biochemical pathway that generates adenosine triphosphate. Because most cellular functions depend on an adequate supply of adenosine triphosphate, CoQ10 is essential for the health of virtually all human tissues and organs. CoQ10 also functions as an antioxidant, assisting in the recycling of vitamin E.42,43
Biosynthesis of CoQ10 begins with the amino acid tyrosine. Pantothenic acid, P5P, and vitamin C are all required for the initial steps in its synthesis. An isoprenyl side chain from farnesyl diphosphate, an intermediate in cholesterol synthesis between 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) and squalene, is then added. An inadequate supply of this intermediate, which can be caused by HMG-CoA reductase inhibitors (cholesterol-lowering drugs of the statin family), results in decreased levels of CoQ10.44
In two of the final steps in the synthesis of CoQ10, methyl groups are provided by SAMe (Figure 54-9). Adequate dietary methionine and a sufficient supply of the nutrients required for the remethylation of homocysteine to methionine (5-methylTHF, methylcobalamin, and betaine) are required to generate sufficient SAMe. Suboptimal amounts of SAMe may negatively affect the body’s ability to synthesize sufficient CoQ10. This relationship between SAMe and CoQ10 has been suggested in various animal studies.45,46
Creatine
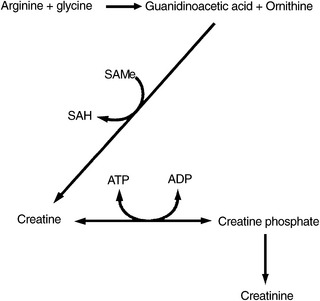
In humans, more than 95% of the total creatine content is located in skeletal muscle, of which approximately one third is in its free form as creatine, also known as methyl-guanidinoacetic acid, whereas the remainder is present in a phosphorylated form as creatine phosphate (also called phosphocreatine). Creatine phosphate is used within skeletal muscle as a means of storing high-energy phosphate bonds.
Creatine is formed in the liver, kidney, and pancreas, beginning with the combination of arginine and glycine to produce guanidinoacetate. A methyl group from SAMe is then transferred, resulting in the formation of creatine. The by-product of this reaction, S-adenosylhomocysteine, is subsequently hydrolyzed into homocysteine and adenosine. To optimize endogenous production of creatine, the amino acids arginine, glycine, and methionine must be available as substrates. Additionally, cofactors needed to optimize remethylation of homocysteine to form methionine are required to recycle the homocysteine to methionine for reuse as SAMe (Figure 54-10). Serum creatine levels have been positively correlated with plasma homocysteine levels (i.e., as creatine levels rise, so do homocysteine levels).47
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


