Gout and Crystal-Induced Arthropathies
Angelo Gaffo
 |
A 65-year-old patient with poorly controlled diabetes, hypertension, heart failure, and a prior diagnosis of gout is hospitalized because of an exacerbation of heart failure with worsening edema and progressive dyspnea. On hospital stay day number 2, symptoms leading to admission were significantly improved. However in the prior 12 hours, he has developed a red, warm, swollen, and extremely tender right ankle.
An arthrocentesis of the affected joint yields cloudy fluid that is positive for the presence of negatively birefringent needle-shaped crystals (Fig. 20.1). A joint glucocorticoid injection was delayed and only analgesic treatment along with low-dose oral colchicine was provided. At 48 hours the synovial fluid culture was reported positive for growth of Klebsiella spp. The patient improved with antibiotic therapy, repeated joint aspirations, low-dose colchicine, and analgesics.
Clinical Presentation
Gout is the clinical manifestation from the tissue deposition of monosodium urate (MSU) crystals. The disease has become more prevalent in Western populations, specifically in certain patient groups such as transplant recipients. It is one of the few medical conditions for which physicians have a nearly complete understanding of the causative and necessary factor for its development, in this case a serum urate concentration above the saturation threshold, or hyperuricemia. This understanding of the etiology and therapeutic target of the disease has not translated into adequate management for the majority of patients with gout because of a combination of factors, including incomplete knowledge of the basic therapeutic principles of the disease and the growing complexity of patients with gout, driven by multiple comorbidities or polypharmacy. Until recently, a scarcity of therapeutic options for gout added to these challenges, but that panorama has started to change.
Epidemiology
Gout is the most common inflammatory arthritis in the United States: according to the most recent estimate by the National Arthritis Data Workgroup, using 1996 data from the National Health Interview Survey (NHIS) and National Health and Nutrition Examination Survey (NHANES), 3.0 million adults older than 18 years had gout in the previous year and 6.1 million adults older than 20 years had gout at some point of their lives. The frequency rates have clearly been increasing in the last decades, with a current estimated prevalence at 940 per 100,000 adults older than 18 years (1).
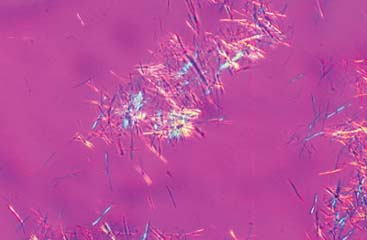
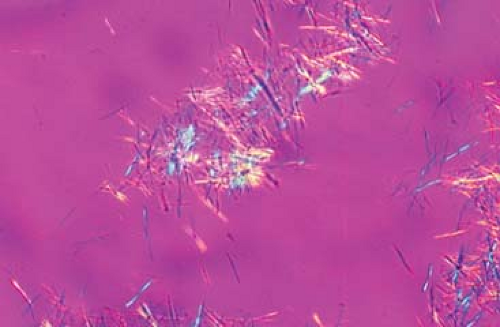 Figure 20.1 Needle-shaped monosodium urate crystals showing characteristic negative birefringence. (Axis of the polarizer points at four O’clock and crystals are predominantly yellow at that direction; perpendicular crystals are predominantly blue.) Courtesy of H. Ralph Schumacher, Jr., M.D., and Janet Dinnella, University of Pennsylvania (http://www.med.upenn.edu/synovium. Accessed June 6, 2011). |
Worldwide, data about measures of disease frequency and time trends are heterogeneous. It is unclear if these variations are because of true differences in frequencies or heterogeneous gout definitions and methods of data collection. The prevalence of gout in the United Kingdom in 1993 seemed to have tripled when compared with that in the 1970s. Other gout high-prevalence populations are the Malayo-Polynesians and New Zealand Maoris (close to 10%). On the other hand, relatively low disease frequencies have been reported in China.
Hyperuricemia
Uric acid, found in serum as urate, is the end product of purine metabolism in humans. The accumulation of urate beyond its solubility point of 6.8 mg/dL defines hyperuricemia, a necessary but not sufficient factor for the development of gout. Gout is the clinical manifestation of the deposition of MSU crystals in tissues.
Clinical Points
Early in the disease course, gout is characterized by acute attacks of arthritis (flares) and asymptomatic intervals. If the disease goes untreated, it morphs into a chronic deforming arthritis with tophi.
Common precipitants of gout flares include acute illnesses, alcohol intake, starvation, excessive intake of purines, and use of certain medications (allopurinol, diuretics, cyclosporine).
Gout flares initially involve the lower extremity joints and peak in intensity within 24 hours.
When starting urate-lowering therapy for gout, it should always be accompanied by prophylactic therapy for gout flares (nonsteroidal anti-inflammatory drugs (NSAIDs), glucocorticoids, or low-dose colchicine).
Calcium pyrophosphate deposition disease can present as acute arthritis (pseudogout), an inflammatory subacute polyarthritis (pseudorheumatoid arthritis), degenerative joint disease (pseudo-osteoarthritis), spinal disease, and a destructive arthritis resembling a neuropathic arthritis.
The importance of hyperuricemia as a causative factor for gout has been corroborated in prospective studies. As part of the Normative Aging Study, a cohort of 2,046 men was followed for 15 years (2). The risk for gout followed a gradient depending on the initial urate level: with an initial level of more than 9 mg/dL, the annual incidence rate was 4.9%. When the initial level was between 7.0 and 8.9 mg/dL, the annual incidence rate was 0.5%, and finally, it was 0.1% with urate levels less than 7.0 mg/dL. The importance of hyperuricemia in predicting gout attacks is limited not only to the initial diagnosis, but also to the management, as it has been demonstrated that low serum urate levels predict freedom from recurrence of gout flares.
Uric acid is synthesized in the liver from purine compounds provided by diet and the endogenous pathway of purine synthesis de novo. It is then released into the circulation almost exclusively in its soluble-form MSU, which is readily available for filtration in the proximal tubules of the kidney. Two mechanisms exist through which an individual could develop hyperuricemia: overproduction (excretion of more than 600 mg/day in the urine while on a purine-free diet, accounting for 10% to 15% of cases) and underexcretion (excretion of less than 400 mg/day, accounting for 85% to 90% of cases). In both cases the problem could be primary (secondary to enzymatic inherited disorders of urate production or defects in renal excretion) or secondary to excessive purine turnover (diet, malignancies), medications, or toxins. For an expanded overview of causes of hyperuricemia, see Table 20.1.
After a nearly complete filtration in the glomerulus, urate undergoes an extensive reabsorption in the proximal tubule largely mediated by a specific organic anion transporter. After the first round of reabsorption, a second cycle of secretion and further reabsorption occurs in the distal portions of the proximal tubules. These final steps determine the net urate excretion, typically 8% to 12% of the initially filtered load.
Once hyperuricemia ensues, the probability of developing gout depends on the concentration of urate in the tissue or joint and other predisposing factors such as a low pH, low temperature, previous trauma to the joint, and lack of mobility (e.g., during sleep, when there is an increased water reabsorption, and concentration of urate). Recent advances in understanding how MSU crystals cause the characteristic gout inflammatory response have been made. Along with other crystals, such as calcium pyrophosphate, silica, and asbestos, MSU is internalized into phagocytes and sensed by the innate immune system as a
danger signal indicative of tissue damage and recognized by a series of sophisticated cytosolic receptors (3).
danger signal indicative of tissue damage and recognized by a series of sophisticated cytosolic receptors (3).
Table 20.1 Causes of Hyperuricemia | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||
The effect of diet as a risk factor for hyperuricemia and gout has been clarified by epidemiologic evidence. Cross-sectional analyses reveal that total beer, liquor, meat, and seafood consumption were associated with higher serum uric acid levels. However, the magnitude of the increase in serum urate in most individuals per unit of intake is relatively small. Wine, total protein, and dairy intake have not been found to be associated with higher serum urate levels. The effect of alcohol intake in inducing flares in patients with established gout is significant.
Fructose intake has gathered much attention as a factor associated with higher levels of serum urate, renal disease, and the development of hypertension. Fructose may induce hyperuricemia through depletion of adenosine triphosphate and its rapid conversion into adenosine monophosphate, which will be later catabolized into uric acid. Epidemiologic studies have established an association between fructose intake and hyperuricemia and gout.
There are several medications and toxins that influence the renal handling of uric acid. Aspirin has a dual effect on serum uric acid levels, with high levels of intake (more than 3 g/day) being uricosuric and lower levels of intake (75 to 2,000 mg/day) promoting uric acid retention. Diuretics (both loop and thiazides) are well known to be associated with higher serum urate levels, possibly through volume contraction and concurrent stimulation of urate reabsorption at the level of the urate absorption receptor in the proximal tubules. Cyclosporine and tacrolimus are widely used drugs for posttransplant immunosuppression and are strongly associated with the development of hyperuricemia and gout;
other chemicals associated with hyperuricemia and gout include lead, pyrazinamide, ethambutol, and niacin.
other chemicals associated with hyperuricemia and gout include lead, pyrazinamide, ethambutol, and niacin.
When approaching patients in which a crystal arthritis is in the differential, a joint aspiration with microscopic examination under polarized light should be performed whenever possible.
Serum urate levels should not be utilized for the evaluation or assessment of suspected gout flares.
As septic arthritis is usually in the differential of crystal arthritides, gram stain and culture of fluid aspirates should usually be performed, even in cases in which crystals are readily identified.
The diagnosis of calcium pyrophosphate deposition disease can be supported by the finding of fine cartilage calcification (chondrocalcinosis) in radiographs of the affected joints.
Examination
Gout is a chronic disease that, if untreated, typically progresses over four phases: (a) asymptomatic hyperuricemia, (b) gout flares, (c) intercritical periods, and (d) chronic, usually tophaceous gout. This progression is illustrated in Figure 20.2. Typically, gout flares present as warmth, swelling, erythema, and pain of abrupt onset in the involved joint, with symptoms peaking over 8 to 12 hours. Patients usually describe the pain as excruciating, and even the slightest physical contact with the affected area (like that produced by a bedsheet) can induce marked suffering. Nighttime onset of symptoms is frequent. Fever, chills, and malaise are commonly part of the presentation in patients with polyarticular flares. In the elderly, atypical presentations that include delirium are frequent. Commonly, the involved joints are in the feet (with the first metatarsophalangeal being eventually involved in more than 90% of cases), ankles, knees, elbows, wrists, and fingers. The predilection for the lower extremities is because of lower temperatures in these joints that favor the precipitation of MSU crystals. Extra-articular sites are also involved, including the bursas (mainly the olecranon and prepatellar bursas) and tendons. The first attack is usually monoarticular, and locates at the first metatarsophalangeal joint in 50% of the cases. Precipitants of gout flares include acute illness (trauma, sepsis, surgery), alcohol intake, starvation, excessive intake of certain food groups (mainly purines), and medications. Drugs such as allopurinol, thiazides, and cyclosporine—which affect serum urate levels or cause body fluid shifts—have been associated with gout flares. Untreated attacks frequently resolve over 3 to 10 days, sometimes with exfoliation of the overlying skin.
The clinical course of gout is characterized by an amelioration or complete disappearance of symptoms during the intercritical period. However, if the disease progresses into chronic gout, the length of these intercritical periods shortens and chronic joint pain persists even during these intercritical periods (Fig. 20.2). It is not infrequent to recover MSU crystals from aspirates of a previously affected joint during the intercritical period.
Chronic gout is characterized by the unremitting nature of the symptoms, destructive arthritis, and the identifiable deposition of solid uric acid in tissues
(tophi). At this stage the involved joints remain persistently uncomfortable, stiff, and swollen. The condition may mimic other inflammatory arthritides, such as rheumatoid arthritis. Superimposed acute flares, which are usually polyarticular and additive, still occur. Tophi appear as a function of the degree and duration of untreated hyperuricemia, usually over extensor surfaces (forearms and the Achilles tendon) and pressure points, typically in the fingers, wrists, knees, and olecranon bursas.
(tophi). At this stage the involved joints remain persistently uncomfortable, stiff, and swollen. The condition may mimic other inflammatory arthritides, such as rheumatoid arthritis. Superimposed acute flares, which are usually polyarticular and additive, still occur. Tophi appear as a function of the degree and duration of untreated hyperuricemia, usually over extensor surfaces (forearms and the Achilles tendon) and pressure points, typically in the fingers, wrists, knees, and olecranon bursas.
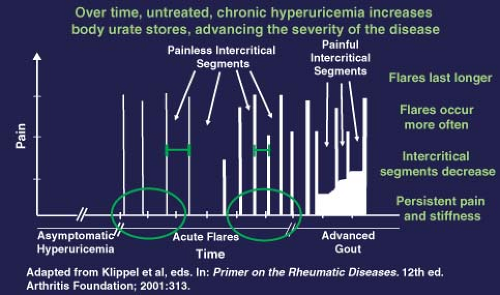 Figure 20.2 Evolution of hyperuricemia and gout. |
Studies
The diagnosis of gout is strongly supported by the combination of a classical clinical presentation (monoarthritis and tophi) along with evidence of hyperuricemia and clinical response to colchicine, NSAIDs, or glucocorticoids. However, it is very important to emphasize that each one of these diagnostic considerations are imperfect and that the diagnosis can only be reliably established by the identification of negatively birefringent needle-shaped MSU crystals from an affected joint or tissue.
The most common way of identifying MSU crystals in patients suspected of having a gout flare is through synovial fluid aspiration from an affected joint. The fluid obtained from joints affected by gout is usually turbid with a yellow tinge, but in extreme cases, it is thick and chalky, with a white coloration. The cell counts are usually in the inflammatory range from 3,000/mm3 up to greater than 50,000/mm3, more than 90% of these cells being polymorphonuclear. Often, needle-shaped MSU crystals can be identified under standard light microscopy. However, the optimal way to visualize MSU is through polarized light microscopy, in which needle-shaped MSU crystals will appear with a bright-yellow or blue coloration (depending on if the axis of the polarizer is parallel or perpendicular to the crystal) against a purple background (Fig. 20.1). Many times the MSU crystals are found inside a leukocyte that is attempting phagocytosis. Despite being the standard way of determining the presence of a gout flare, synovial fluid analysis with crystal identification has some drawbacks. For example, patients with gout and hyperuricemia that are not having a gout flare could have MSU crystals in their joint synovial fluid (usually in the context of a noninflammatory cell count). Also, the aspiration of small joints could be technically challenging and the procedure could be difficult to perform for untrained practitioners. Another potential issue is that synovial fluid has to be promptly analyzed as cells counts decline and crystals degenerate when sample processing is delayed. Finally, the identification of MSU crystals in partially treated patients or those in which the gout flare is resolving can be challenging and requires lots of patience on part of the examiner. Bursal fluid, tophi aspirates, and tissue samples can also be analyzed with the purpose of identifying MSU crystals.
Measurement of serum urate is an unreliable predictor of gout flares and should not be used with diagnostic purposes as up to 40% of cases of acute gout occur in the setting of normouricemia. On the other hand, hyperuricemia is frequent in the general population and could be present in the setting of an acute arthritis secondary to rheumatoid disease, psoriasis, infection, and so on. Other ancillary investigations such as the measurement of urine urate excretion and plain radiographs have a limited role in diagnostic and management decisions.
The American College of Rheumatology (formerly American Rheumatism Association) proposed preliminary criteria for the diagnosis of acute gouty arthritis in 1977 (Table 20.2; 4). Despite their widely adopted use and citation, those were never validated, and important limitations in their performance have been recognized. More recently, the European League Against Rheumatism has proposed recommendations for the diagnosis of gout that translate
into a diagnostic rule (5). In addition, a Dutch primary care group developed a diagnostic rule for identification of gout in patients with monoarthritis without the need of an arthrocentesis that could be of great usefulness to clinicians in communities without access to prompt synovial fluid analysis (Table 20.2; 6).
into a diagnostic rule (5). In addition, a Dutch primary care group developed a diagnostic rule for identification of gout in patients with monoarthritis without the need of an arthrocentesis that could be of great usefulness to clinicians in communities without access to prompt synovial fluid analysis (Table 20.2; 6).
Table 20.2 Rules for the Classification and Diagnosis of Gout
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|
|---|





