Chapter 4 Genetics of Human SLE
Monogenic Deficiencies and Rare Mutations with SLE
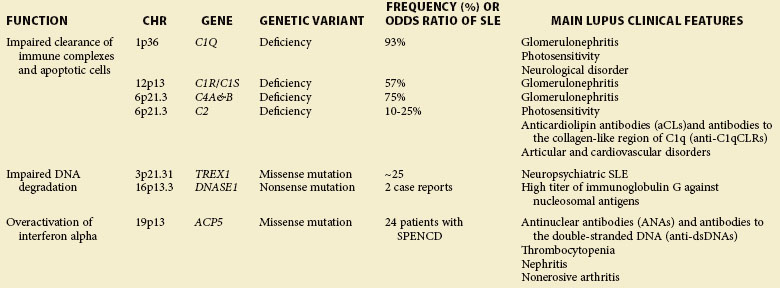
Most patients affected with SLE have no family history of this disease. In families with multiple affected members, the disease occurrence does not follow the classic mendelian inheritance model for a single-gene disorder. However, in a few cases, SLE is associated with rare but highly penetrant mutations (Table 4-1), resulting in deficiency of classical complement components and/or defective degradation of DNA.
Complement Deficiency
An extremely strong genetic risk for SLE is conferred by a complete deficiency in one of the classical complement pathway genes, such as C1Q, C1R/S, C2, C4A, and C4B, even though these deficiencies are relatively rare. The incidence of SLE or lupus-like manifestations has been identified in 93% of homozygous C1Q-deficient individuals, in 57% of C1R/C1S-deficient individuals, in 75% of C4-deficient individuals, and in 10% to 25% of C2-deficient individuals.1 Patients with SLE and deficiency of C1Q or C4 usually demonstrate disease at a young age without a female predominance and have an approximate 30% frequency of renal involvement (glomerulonephritis).1 In contrast, patients with SLE and C2 deficiency show a sex distribution similar to that seen in lupus in general (female/male 7 : 1) and demonstrate disease later in life.2 The severity of disease in patients with SLE and C2 deficiency does not differ from that in most patients with SLE; however, an increased rate of skin or cardiovascular involvement and a low frequency of glomerulonephritis is observed in C2 deficiency (reviewed by Jonsson3). Complement is critical for the opsonization and clearance of autoantibody-containing immune complexes (ICs). Deficiencies of complement components in the classical pathway are involved in several key steps in the SLE; pathogenesis, including reduced tolerance of autoantigens, reduced handling of apoptotic cell debris and IC clearance, and dysregulation of TLR (Toll-like receptor)– or IC-induced cytokines.1
TREX1
Mutations in one of three genes encoding the intracellular nucleases, TREX1 (a major 3′-5′ DNA exonuclease), RNase H2 (degrades DNA : RNA hybrids), and SAMHD1 (a putative nuclease), cause the Aicardi-Goutières syndrome (AGS), which shares several features with SLE, such as hypocomplementemia and antinuclear autoantibodies.4 Of note, missense mutations of TREX1 are found in 0.5% to 2.7% of patients with SLE but are nearly absent in healthy controls.5,6 A 2011 analysis of more than 8000 multiancestral patients with SLE has revealed a risk haplotype of TREX1 associated with neurologic manifestations, especially seizures, in patients of European descent.6 TREX1 serves as a cytosolic DNA sensor, preferentially binds to single-stranded DNA, and functions as a DNA-degrading enzyme in granzyme-A–mediated apoptosis.7 TREX1 deficiency impairs DNA damage repair, leading to the accumulation of endogenous retroelement-derived DNA. Defective clearance of this DNA induces IFN production of interferon (IFN) and an immune-mediated inflammatory response, promoting systemic autoimmunity.7
TRAP
The immuno-osseous dysplasia spondyloenchondrodysplasia (SPENCD) has been regarded primarily as a skeletal dysplasia, but patients with the disease also show a high frequency of autoimmune phenotypes, including SLE, Sjögren syndrome, hemolytic anemia, thrombocytopenia, hypothyroidism, inflammatory myositis, Raynaud disease, and vitiligo.8,9 Loss-of-function mutations in the acid phosphatase 5 gene (ACP5; encoding tartrate-resistant acid phosphatase, TRAP), which have been identified as causative of the disease,8,9 result in an elevated serum IFN-α activity and an IFN signature in patients with SPENCD.8 Because TRAP is responsible for dephosphorylating osteopontin (OPN; encoded by SPP1), a multifunctional cytokine involved in immune system signaling, it is possible that in the absence of TRAP, OPN would remain phosphorylated and maintain persistent activation of IFN-α through the TLR9/MyD88 pathway.9 Of interest, SPP1 genetic polymorphisms have been associated with SLE and enhanced IFN-α activity, and elevated OPN protein values are correlated with the inflammatory process and SLE development.10,11
DNASE 1
Deoxyribonuclease I (DNase I, encoded by DNASE1) is a specific endonuclease facilitating chromatin breakdown during apoptosis. DNase I activity is important to prevent immune stimulation, and reduced activity may result in an increased risk for production of antinucleosome antibodies, a hallmark of SLE.12 Several studies have found a connection between low DNase I activity and the development of human or murine SLE.13,14 By sequencing the DNASE1 gene in 20 Japanese patients with SLE, Yasumoto15 found two female patients with a mutation in exon 2; the mutation resulted in a replacement of lysine with a stop signal, so they had decreased DNase I activity and an extremely high immunoglobulin G (IgG) titer against nucleosomal antigens.15 Although this mutation has not been confirmed in other patient populations, specific common single-nucleotide polymorphisms (SNPs) of DNASE1 (e.g., Q244R) have been associated with SLE susceptibility but not with DNase I activity nor with autoantibody titers.16,17
Polygenic Common Variants in SLE
Genome-Wide Linkage Studies
Linkage analysis is a comprehensive and unbiased approach, in which a few hundred genetic markers (such as DNA polymorphisms) are screened at 10- to 15-kb (kilobase) genomic intervals to identify chromosomal regions cotransmitted with disease in families containing multiple affected members. A total of 12 genome-wide scans and eight targeted linkage analyses have established 9 loci reaching the threshold for significant linkage to SLE (1q23, 1q31-32, 1q41-43, 2q37, 4p16, 6p11-21, 10q22-23, 12q24, and 16q12-13).18 An alternative approach, that of stratifying by the presence of a clinical symptom in multiplex pedigrees, has led to the identification of 11 significant loci linked to particular SLE manifestations (reviewed by Sestak18). Progress toward further localizations of underlying causal variants has met with limited success because linkage intervals usually span large genomic regions that contain hundreds, if not thousands, of potential candidate genes, and because some important genes (e.g., IRF5) associated with SLE are not located within established linkage regions.
Candidate Gene Studies
Candidate gene studies are traditionally used to assess whether a test genetic marker (usually SNPs are under investigation) is present at a higher frequency among patients with SLE than in ethnically matched healthy control individuals. Candidate genes are chosen on the basis of either their functional relevance to the disease pathogenesis or their locations within chromosomal regions implicated in linkage studies. The test SNP observed with greater than expected frequency in individuals with disease is either a functional, disease-causing variant (a direct association) or a nonfunctional variant that exhibits strong linkage disequilibrium (LD) with the functional variant (an indirect association).19 Literally hundreds of association studies of SLE were published in the last century, which, however, uncovered a limited number of confirmed SLE susceptibility genes because of small sample collections and/or a lack of dense marker coverage (reviewed by Tsao20). These limitations in linkage and candidate gene studies have hindered our understanding of the pathways causally involved in disease pathogenesis. This situation changed dramatically with the advent of the GWAS.
Genome-Wide Association Studies
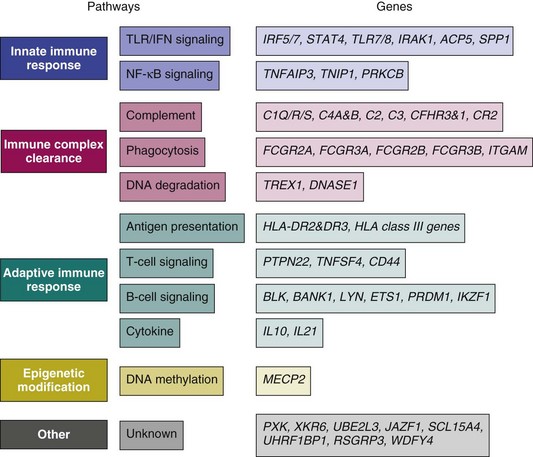
The GWAS, an important step beyond the two previously mentioned methods, is built on efforts to identify associations of common genetic variations across the entire human genome with disease susceptibility. Rapid advances in technology have enabled a simultaneous genotyping of up to 1 million SNPs in a single GWAS. A typical GWAS usually consists of the following four parts21: (1) selection of a large number of individuals with disease of interest and a well-matched comparison group, (2) genotyping and data review to ensure high genotyping quality, (3) statistical association tests of the SNPs passing quality thresholds, and (4) replication of identified associations in an independent population or assessment of their functional implications. Since 2007, six GWASs22–27 and a series of subsequent large-scale replication studies in SLE using both European and Asian populations not only have confirmed associations at previously established loci but also, and more importantly, have identified a number of novel loci (Table 4-2). Many of the disease-specific genes can be grouped into three major immunologic pathways (Figure 4-1). A growing number of genes seem to predispose to multiple autoimmune disorders, including SLE, rheumatoid arthritis (RA), systemic sclerosis (SSc), type 1 diabetes (T1D), Crohn disease (CD), Graves disease (GD), and psoriasis, highlighting the shared immunologic mechanisms conferred by common genetic variants among some of these disease processes. A few genes that cannot be mapped to a known disease pathway are likely to reveal new paradigms for disease pathogenesis and may provide new therapeutic targets for disease management.
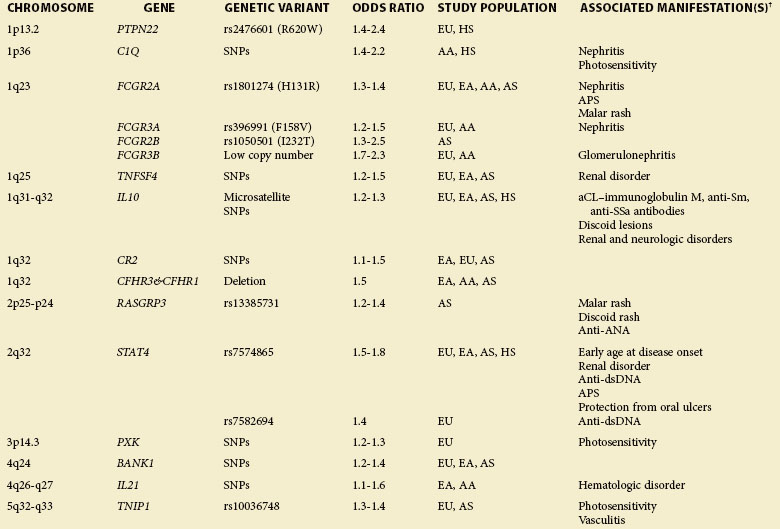
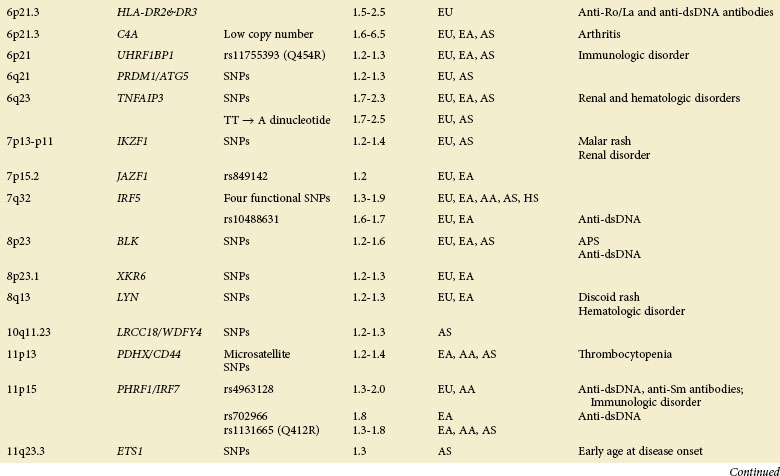
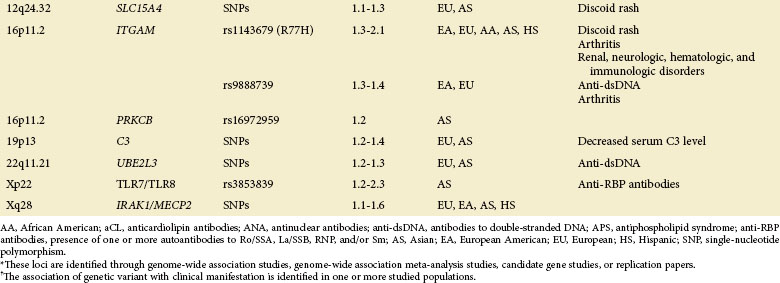
A role for gene copy number variation (CNV) in SLE has been appreciated through studies of the complement component 4 (C4), Fcγ receptor IIIB (FCGR3B), TLR 7 (TLR7), and later work in complement regulator factor H–related 3 and 1 (CFHR3 and CFHR1).28,29 CNVs can be detected either through direct scoring or identification of SNP markers known to be in LD with CNVs. The availability of large SNP-based GWAS datasets and future genetic screens using more dense markers, including structural variants (known as CNVs), will facilitate the genome-wide analysis and identification of CNVs predisposing to SLE susceptibility.
Human Leukocyte Antigen
Major Histocompatibility Complex Structure
The classical major histocompatibility complex (MHC) (also referred as the human leukocyte antigen [HLA]) region encompasses approximately 3.6 Mb on 6p21.3 and is divided into the class I (telomeric), class III, and class II (centromeric) regions. The class I and class II regions encode the classical HLA genes (HLA-A, -B, -C, -DR, -DQ, and –DP) involved in antigen presentation to T cells and transplant compatibility. The class I and class II molecules are the most polymorphic human proteins known to date. Because these molecules shape the immune repertoire of an individual, the extreme polymorphism is thought to have evolved in response to infectious pathogens. Perhaps that is the reason that the MHC is associated with more diseases than any other region of the human genome and is linked to most, if not all, autoimmune disorders. The class III region lies between the class I and class II regions and is the most gene-dense region in the genome, encoding a variety of molecules including the early complement components (e.g., C2, C4, and factor B), cytokines (e.g., tumor necrosis factor alpha [TNF-α] and lymphotoxin-α), the heat shock protein cluster, and proteins involved in growth and development. Given the existence of long-range LD- and MHC-related genes outside this classically defined locus, there comes to be a concept of the extended MHC (xMHC), spanning nearly 7.6 Mb of the genome, that consists of five subregions: the extended class I subregion (HIST1H2AA to MOG; 3.9 Mb), classical class I subregion (C6orf40 to MICB; 1.9 Mb), classical class III subregion (PPIP9 to NOTCH4; 0.7 Mb), classical class II subregion (C6orf10 to HCG24; 0.9 Mb); and extended class II subregion (COL11A2 to RPL12P1; 0.2 Mb);30 Of the 421 genes within this extended region, 60% are expressed and approximately 22% have putative immunologic function.
HLA Class II Region and SLE
The association between SLE and variations in the HLA region has been extensively studied. Until 2005, most published disease association studies of HLA using small case-control panels of predominant European ancestries were restricted to a subset of about 20 genes, including the classical HLA loci (HLA-A, -B, -C, -DRB, -DQA, -DQB, -DPA, -DPB), TNFA, LTA, LTB, TAP, MICA, MICB and the complement loci (C2, C4A, C4B, and CFB) (reviewed by Fernando30). A pooled analysis of the past 30 years of research work regarding HLA genetics in SLE has pointed to the most consistent association with HLA-DR3 (or DRB1*0301; one of the alleles from the previous DR3 specificity) and HLA-DR2 (or DRB1*1501; one of the alleles from the previous DR2 specificity) and their respective haplotypes in predominantly European-derived populations.31 In particular, the strongest associations were for the HLA-DR3 haplotypes, B8-DRB1*0301 and B18-DRB1*0301, with odds ratios (ORs) ranging from 1.5 to 2.5; whereas the associations of DR2, DR15, DRB1*1501, and DQB1*0602, which mapped to the DR2/DRB1*1501 haplotype, exhibited an OR of 1.7.31 Studies in non-European populations have revealed inconsistent results. For instance, the association with another HLA-DR2 subtype, DRB1*1503, was only found in African Americans, who demonstrated no association with DR2 or DR3 alleles.32 HLA-DRB1*1602 has been observed in Mexican Mestizo, Thai, and Bulgarian populations; and HLA-DRB1*0401 has been seen largely in Mexican Mestizo and Hispanic populations.31 Two further class II alleles, HLA-DQA1*0401 and HLA-DQB1*0402, reside on a DR8 haplotype that is uncommon in European populations.33
Given the role for HLA class II molecules in T cell–dependent antibody responses, there is a close association of class II alleles, especially HLA-DR and HLA-DQ alleles with autoantibody subsets in patients with SLE of multiple ancestries (reviewed by Fernando30). The strongest associations have been demonstrated between anti-Ro/La antibodies and DR3 and DQ2 (DQB1*0201), which are in strong LD. Predominant associations with antiphospholipid antibodies—including anticardiolipin antibody (aCL), lupus anticoagulant (LA), and anti-β2 glycoprotein I antibody (anti-β2GPI—are found for the DR4 (DRB1*04)/DQ8 (DQB1*0302) haplotype and other class II alleles. The HLA associations with other autoantibodies, including anti–double-stranded DNA (anti-dsDNA), anti-RNP, and anti-Sm, are much more complex, yielding inconclusive results.
HLA Class III Region and SLE
Despite a remarkably high gene density in the HLA class III region, only complement C4 CNVs and polymorphisms of tumor necrosis factor (TNFA) have been studied in detail in SLE (reviewed by Wu34 and Postal35). It is concluded that a lower copy number of C4 (due to increases in homozygous and heterozygous deficiencies of C4A but not C4B) increases risk and a higher copy number decreases risk for SLE. CNVs of C4 genes determine the basal levels of circulating complement C4 proteins that function in the clearance of ICs, which can otherwise promote autoimmunity. Studies of TNFA polymorphisms have pointed to the promoter SNP-308A/G for its association with SLE either independently or as a part of an extended HLA haplotype, HLA-A1-B8-DRB1*0301-DQ2, in multiple ancestries. However, this association is not confirmed in other similar studies, so additional work is needed to clarify the role of genetic variants of TNFA in susceptibility to SLE.
With high-density genetic markers, GWASs and fine-mapping studies of SLE in populations of European and Asian ancestries have revolutionized our understanding of the HLA genetic contributions, which not only confirm predominant association signals at the class II region but also highlight the importance of class III genes in SLE susceptibility. For example, one SNP (rs3131379) of the HLA class III locus MSH5 (mutS homolog 5) exhibited the highest association in a GWAS conducted in 2008.23 A mapping study in 314 European families with SLE reported two distinct and independent signals36: one from a small, 180-kb class II region tagged by HLA-DRB1*0301 allele and the other observed at an SNP marker (rs419788) in the class III gene SKIV2L (superkiller viralicidic activity 2–like [Saccharomyces cerevisiae]). Examination of LD structure around this marker (rs419788) showed this class III signal to be restricted to a 40-kb interval containing the genes CFB, RDBP (RD RNA binding protein), DOM3Z (dom-3 homolog Z [Caenorhabditis elegans]), and STK19 (serine-threonine kinase 19). CFB encodes complement factor B, which is a vital component of the alternate complement pathway. The functions of RDBP, SKIV2L, DOM3Z, and STK19 are not well characterized, although their products have been reported to play a role in messenger RNA (mRNA) processing. Of note, this study provided evidence against an independent effect of TNFA-308G/A polymorphism in SLE, which is inconsistent with results from another meta-analysis study.37 Another collaborative study in multiple immune-mediated diseases indicated that the highest association signal for SLE was detected at SNP (rs1269852), located in the class III region between TNXB (tenascin XB) and ATF6B (activating transcription factor 6 beta) genes.38 Other class III association signals were peaks centered on the NOTCH4 gene and those on either side of the RCCX module (which contains C4A and C4B genes along with three neighboring genes). The influence of CNVs at the complement C4/RCCX locus in relation to the association signals revealed in this study remains to be established.38
Innate Immunity Genes
IRF5
IRF5 encodes for interferon regulatory factor 5 (IRF5), a pivotal transcription factor in the type I IFN pathway that regulates the expression of IFN-dependent genes, inflammatory cytokines, and genes involved in apoptosis. IRF5 is one of the most strongly and consistently SLE-associated genes outside the HLA region, conferring a modest risk with an OR of 1.3 or more. Predominant associations of IRF5 with SLE in populations of multiple ancestries are identified at four functional variants, a 5–base pair (bp) indel (insertion/deletion) near the 5′ untranslated region (UTR) rs2004640 in the first intron, a 30-bp indel in the sixth exon, and rs10954213 in the 3′ UTR.39 Alleles of these functional variants in different combinations define various haplotypes that are associated with increased, decreased, or neutral levels of risk for SLE. The risk haplotypes have functional consequences, including greater expression of IRF5 mRNA and IFN-inducible chemokines, as well as elevated IFN-α activity.40,41 Indeed, a critical role for IRF5 in mediating lupus pathogenesis is demonstrated in murine models of lupus-like disease using Irf5-deficient and Irf5-sufficient FcγRIIB−/− Yaa mice42 or Irf5−/− MRL/lpr mice.43
STAT4
The signal transducer and activator of transcription 4 (encoded by STAT4) can transmit signals from the receptor for type I IFN, interleukin (IL) 12, and IL-23, and contribute to autoimmune responses by affecting the functions of several innate and adaptive immune cells. The SLE-associated SNP (rs7574865) in the third intron of STAT4 was first identified in several case-control studies, exhibiting an OR of 1.5 to 1.7,44 and was confirmed by GWASs using populations of European or Asian ancestry.23,25–27,45 The risk allele of rs7574865 is associated with a more severe SLE phenotype, characterized by development of disease at an early age (<30 years), a high frequency of nephritis, the presence of antibodies against dsDNA, and an increased sensitivity to IFN-α signaling.46–48 Fine-mapping studies led to the identification of several markers that are independently associated with SLE and/or with differential levels of STAT4 expression,47,49,50 and a 73-kb risk haplotype common to European Americans, Koreans, and Hispanic Americans.50
PHRF1/IRF7
Two independent studies in European populations have reported an SLE-associated SNP (rs4963128) in a gene of unknown function named PHD and RING-finger domains 1 (PHRF1, also known as KIAA1542).23,45 Given that a strong LD (r2 = 0.94) between this disease-associated SNP and the 3′UTR PHRF1 SNP (rs702966) is within a 0.6-kb flanking region of the IRF7 gene, this observed association might be attributable to its close proximity to IRF7 (which codes interferon regulatory factor 7).23 Like IRF5, IRF7 is a transcription factor that can activate transcription of IFN-α and IFN-α–inducible genes downstream of endosomal TLRs. Two studies support PHRF1/IRF7 as an SLE susceptibility locus with the following findings: (1) patients with SLE carrying the risk allele of PHRF1 SNP (rs702966) and expressing autoantibodies to dsDNA or Sm exhibit elevated serum IFN-α activity51 and (2) the major allele of a nonsynonymous SNP (Q412R) in IRF7 confers elevated IFN-stimulated response in vitro and predisposes to SLE in Asians, European Americans, and African Americans.52 However, a complete assessment of this locus with dense genetic markers and/or sequencing to localize all possible causal variants is still pending.






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


