Genetic Aspects of Orthopaedic Conditions
José A. Morcuende
Benjamin A. Alman
A genetic and molecular revolution is happening in medicine. Led by the Human Genome Project, genetic information and concepts are changing the way disease is defined, diagnosis is made, and treatment strategies are developed. The profound implications of actually understanding the molecular abnormalities of many clinical problems are affecting virtually all medical and surgical disciplines. Importantly, genetic technologies will increasingly drive biomedical research and the practice of medicine in the near future.
It is important for those interested in the musculoskeletal system to be aware of the genetic cause of its inherited disorders in order to make appropriate referrals for genetic counseling and to refine the prognosis and natural history in each individual patient. Current management revolves around treatment to prevent or minimize medical complications, psychosocial support of patients and their families, and modification of the environment where appropriate. Gene discoveries will allow the development of tests to detect disease or to quantify the risk of disease. Furthermore, applying this knowledge is the best hope for developing strategies to modify the pathologic effect of the gene (drug therapy) or repair the gene (gene therapy) or for approaches to restore lost or affected tissue (tissue engineering). Instead of an empiric trial-and-error approach to therapy, it may become feasible to tailor treatment to the specific molecular malfunction. No case demonstrates better this evolution than Marfan syndrome where the discovery of the mutated gene (fibrillin-1) led to a search for the pathophysiologic involvement of transforming growth factor β (TGF-β) signaling (abnormal levels of activation) and ways to treat it (with angiotensin II receptor blockers), now in clinical trials (1, 2 and 3).
Given the large number of inherited musculoskeletal abnormalities and the power and speed of current genetic and developmental biology information, a few selected disorders are discussed in this chapter to illustrate specific concepts on the basic genetic concepts. It also discusses current classifications and clinical evaluation and provides a perspective about genetic counseling.
MOLECULAR BASIS OF INHERITANCE
It is estimated that there are more than 10 million living species on Earth today. Each species is different, and each reproduces itself faithfully: parent organisms hand down information specifying, in extraordinary detail, the characteristics that the offspring shall have. This phenomenon of heredity is central to the definition of life. Hereditary phenomena have been of interest to humans long before biology or genetics existed as a scientific discipline. Ancient peoples improved crops and domesticated animals by selecting desirable individuals for breeding. However, the prevailing notion at the time was that the spermatozoon and the egg contained sampling of essences from the various parts of the parental bodies and at conception, they blended together. But there were many instances in which this mode of inheritance could not explain many of the observations on heredity. As a modern discipline, genetics began in the 1860s with the work of Gregor Mendel who performed a set of experiments that pointed to the existence of biological elements that we now call genes.
Genetics is the study of genes at all levels, from molecules to populations. The discovery of genes and the understanding of their molecular structure and function have been a source of profound insight into two of the biggest mysteries of biology: what makes a species what it is and what causes variation within a species? In addition, it has exploded our understanding of human diseases, specifically genetic disorders. Genetics took a major step forward with the notion that the genes, as characterized by Mendel, are part of specific cellular structures, the chromosomes. This major concept has become known as the chromosome theory of heredity. This fusion between genetics and cell biology is still an essential part of genetic analysis today and has tremendous implications in many fields including medical genetics (4, 5 and 6).
Genes encode for the amino acid sequence of the proteins that are the main determinants of the properties of an organism (and also for single catalytic or structural RNAs that do not code for proteins but are essential for RNA metabolism). Importantly, any one gene can exist in several forms
(called alleles) that differ from each other, generally in small ways. Allelic variation causes hereditary variation within a species. Three fundamental properties are required of genes: (a) replication—hereditary molecules must be capable of being copied to ensure the continuation of a species from one generation to the next; (b) information storage—genes have the essential coding specifications to be translated to proteins and their by-products which constitute the building blocks of the organism; and (c) mutation—genes can change over time and this is the basis for variation within species and for evolution.
(called alleles) that differ from each other, generally in small ways. Allelic variation causes hereditary variation within a species. Three fundamental properties are required of genes: (a) replication—hereditary molecules must be capable of being copied to ensure the continuation of a species from one generation to the next; (b) information storage—genes have the essential coding specifications to be translated to proteins and their by-products which constitute the building blocks of the organism; and (c) mutation—genes can change over time and this is the basis for variation within species and for evolution.
Chromosomes.
A cell’s basic complement of DNA is called its genome, and it carries the information for all the proteins the organism will ever synthesize. The body cells of most animals contain two genomes, that is, they are diploid. The cells of most bacteria, algae, and fungi contain just one genome, that is, they are haploid. Interestingly, even closely related species with similar genome sizes can have very different numbers and sizes of chromosomes. Thus, there is no simple relationship between chromosome number, species complexity, and total genome size.
The genome itself is made up of extremely long molecules of DNA that are organized into chromosomes. Karyotype refers to the complement of chromosomes as visualized by cytogenetic analysis at mitosis. Human somatic cells contain two sets of 23 chromosomes, for a total of 46, referred to as euploidy. The maternal and paternal chromosomes of a pair are called homologous chromosomes (homologs). The only nonhomologous chromosome pair is the sex chromosomes, where Y is inherited from the father and X from the mother (Fig. 2-1).
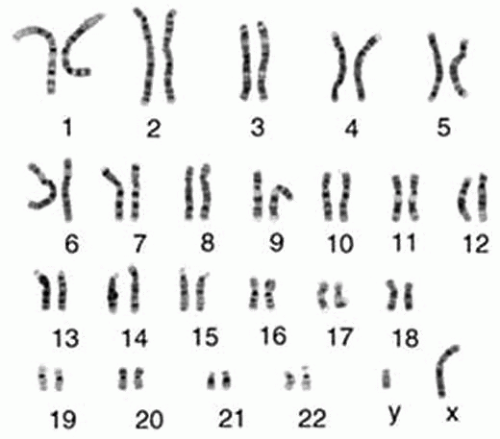 FIGURE 2-1. Banding patterns of human chromosomes. The display of the 46 human chromosomes at mitosis is called the human karyotype. Chromosomes 1 to 22 are numbered in approximate order of size. A typical human somatic cell contains two of each of these chromosomes, plus two sex chromosomes—two X chromosomes in a female and one X and one Y in a male. |
Each chromosome carries a different array of genes linearly arranged along the chromosome (each gene occupies a particular position or locus); therefore, genes are present twice (one allele coming from the mother and one from the father). Alleles are truly different versions of the same basic gene. Looked at another way, gene is the generic term and allele is the specific. Allelic variation is the basis for hereditary variation and genetic disorders (7) (Fig. 2-2).
Somatic cells divide by the process of mitosis. When a cell divides, all chromosomes are replicated and each daughter cell contains the full complement of chromosomes. When a chromosome is replicated, all the genes in that chromosome are automatically copied along with it. In contrast, germ-line cells undergo meiosis during which the diploid number of 46 chromosomes is reduced to the haploid number of 23, including one of each of the autosomes and either the sex chromosome X or Y. Therefore, gametes are haploid, containing one chromosome set.
The highly programmed chromosomal movements in meiosis cause the equal segregation of alleles into the gametes. For instance, during meiosis in a heterozygote A/a, the chromosome carrying A is pulled in the opposite direction from the chromosome carrying a, so half the resulting gametes carry A and the other half carry a. The random assortment of each of the chromosome pairs during meiosis is central to the inheritance pattern of single-gene disorders and some forms of chromosomal derangements. In addition, gene pairs on separated chromosome assort independently at meiosis.
The force pulling the chromosomes to cell poles is generated by the nuclear spindle, a series of microtubules made of the protein tubulin. Microtubules attach to the centromeres of chromosomes by interacting with another specific set of proteins located in this area. The orchestration of these molecular interactions is complex, yet constitutes the basis of the laws of hereditary transmission in eukaryotes.
Gene Structure and DNA Replication.
We have discussed how genes are arranged in chromosomes, but to form a functional chromosome, a DNA molecule must be able to replicate and the copies must be separated and reliably partitioned into daughter cells at each cell division. This process occurs through an ordered series of stages, collectively known as cell cycle.
Approximately 30,000 genes are present in the human genome. Genes are made of linearly aligned nucleotides along the DNA molecules. DNA is a linear, double-helical structure looking rather like a molecular spiral staircase. It is composed of two intertwined chains of building blocks called nucleotides. Each of the four nucleotides is usually designated by the first letter of the base that it contains: A (adenine), G (guanine), C (cytosine), or T (thymine). The nucleotides are connected to each other at the 3’ and 5’ position; hence each DNA chain is said to have a polarity and each chain runs in the opposite direction, that is, the chains are said to be antiparallel. The two nucleotide chains are held together by hydrogen bonds between the nucleotide bases. The bases that form base pairs (A-T and G-C) are said to be complementary.
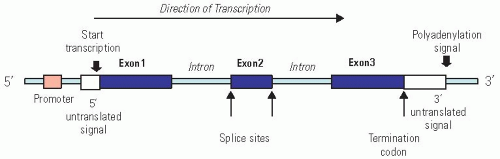 FIGURE 2-2. Generalized structure of a human gene. This example has three exons (coding sequences) and two introns. Note the promoter as the regulatory site for initiation of transcription, the splice sites and termination codon used for mRNA processing. RNA splicing regulation can generate different versions of a protein in different cell types. |
The biological role of most genes is to carry or encode information on the composition of proteins. The protein composition, together with the timing and amount of production of each protein, is an extremely important determinant of the structure and physiology of the organism. Each gene is responsible for coding for one specific protein or a part of a protein. In all cells, the expression of individual genes is regulated: instead if manufacturing its full repertoire of possible proteins at full tilt all the time, the cell adjusts the rate of individual gene transcription and translation independently, according to need and the environment (Fig. 2-3). Regulation of gene expression determines the unique biologic qualities of each cell or its phenotype.
Structurally, a typical gene, defined as a segment of DNA that specifies a functional RNA, is composed of (a) a regulatory region called promoter to which various proteins bind, causing the gene to be transcribed at the right time and in the right amount; (b) a region at the end containing sequences encoding the termination of transcription; and (c) the coding region, which is divided into smaller pieces called exons that are separated by noncoding regions called introns (whose function is still unclear). Each set of three nucleotides in the coding region of the exons is called a codon, and the sequence of codons specifies the amino acid composition and order in the protein.
Transcription and Ribonucleic Acid Processing.
The first step taken by the cell to make a protein is to copy, or transcribe, the information encoded in the DNA of the gene as a related but single-stranded molecule called RNA. This primary RNA copy made from the gene is called a primary transcript and represents a “working copy” of the gene. Whereas the cell’s genetic information archive in the form of DNA is fixed and sacrosanct, the RNA transcripts are mass-produced and disposable. Thus, the primary role of the transcripts is to serve as intermediaries in the transfer of genetic information: they serve as messenger RNA (mRNA) to guide the synthesis of proteins according to the genetic instructions stored in the DNA.
The primary RNA contains both introns and exons; therefore, to make it functional, it needs to be further processed. After the introns are spliced out (RNA splicing), the remaining exons form the protein-encoding continuous sequence called mRNA. This mRNA molecule is transposed from the nucleus to the cytoplasm where it will be translated to protein by the ribosome machinery. These processing steps can critically change the “meaning” of an RNA molecule and are therefore crucial for understanding how eukaryotic cells read the genome. Interestingly, a cell can splice the primary RNA in different ways and thereby make different chains for the same gene—a process called alternative RNA splicing. It is estimated that at least one-third of the human genes produce multiple proteins this way. When different splicing possibilities exist at several positions in the transcript, a single gene can produce dozens of different proteins. Furthermore, the regulation of RNA splicing can generate different versions of a protein in different cell types, according to needs of the cell. Finally, there is also the mechanism of RNA editing which alters the nucleotide sequences of mRNA transcripts once they are transcribed.
Some protein-encoding genes are transcribed more or less constantly; they are the “housekeeping” genes that are always needed for basic cellular functions. Other genes may be rendered unreadable or readable to suit the cellular functions at particular moments and under particular external conditions. Regulation of gene expression is thought to be at multiple levels, although control of the initiation of transcription
(transcriptional control) usually predominates. Some genes, however, are transcribed at constant level and turned on and off solely by posttranscriptional regulatory processes. Most of these control process require the recognition of specific sequences or structures in the RNA molecule being regulated. This recognition is accomplished by either a regulatory protein or a regulatory RNA molecule.
(transcriptional control) usually predominates. Some genes, however, are transcribed at constant level and turned on and off solely by posttranscriptional regulatory processes. Most of these control process require the recognition of specific sequences or structures in the RNA molecule being regulated. This recognition is accomplished by either a regulatory protein or a regulatory RNA molecule.
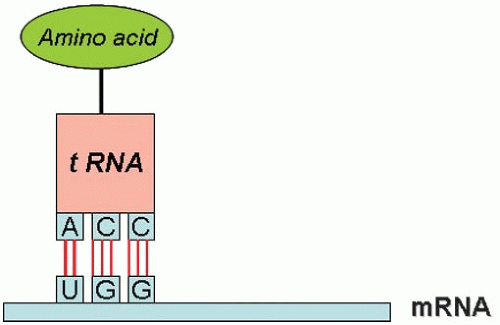 FIGURE 2-3. The process of translation converts genetic information into amino acid sequence. This process takes place on the ribosomes, and each amino acid is brought to the ribosome by a specific tRNA molecule docking at a specific codon of the mRNA. |
Translation.
The process of reading the genetic information of the mRNA sequence and converting it into an amino acid sequence is called translation (rather like converting one language (genotype) into another (phenotype). This process takes place on the ribosomes and each amino acid is brought to the ribosome by a specific transfer RNA (tRNA) molecule that docks at a specific codon of the mRNA. The ribosome is a giant multimolecular machine formed of two main chains of RNA, called ribosomal RNA (rRNA), and more than 50 different proteins. Mechanistically, a ribosome attaches to the 5’ end of an mRNA molecule and moves along the mRNA, catalyzing the assembly of the string of amino acids that will constitute the primary structure of the protein. The first codon of the coding sequence is methionine, and the primary chain is called a polypeptide. At the end of the mRNA, a termination codon causes the ribosome to detach and recycle to another mRNA. Trains of ribosomes pass along an mRNA molecule, each member making the same type of polypeptide.
Protein Assembly.
There are four levels of organization in the structure of a protein. The amino acid sequence is known as the primary structure of the protein. Stretches of polypeptide chain that form α helices and β sheets constitute the secondary structure. The full tridimensional organization is referred as the tertiary structure. Finally, when a protein is formed as a complex of more than one polypeptide chain, the complete structure is designated the quaternary structure (with each polypeptide chain called a protein subunit).
Studies of the conformation, function, and evolution of proteins have revealed a unit of central importance distinct from the previously described organization. This is the protein domain. This is a substructure produced by any part of a polypeptide chain that folds independently into a compact, stable structure. The different protein domains are often associated with different functions.
Proteins are important either as structural components— such as collagen for bone and cartilage—or as active agents in cellular processes, for example, enzymes and active transport proteins. In addition, many proteins undergo numerous posttranslational modifications such as terminal excisions or enzymatic modifications adding sugars, or are assembled into complex polymers.
Interestingly, the synthesis of proteins encoded by genes on the mitochondria takes place on ribosomes inside the organelles themselves. Therefore, the proteins in mitochondria are of two different origins: either encoded in the nucleus and imported into the organelle or encoded in the organelle and synthesized within the organelle compartment. This fact is important since some genetic disorders are now recognized originating in mutations of these mitochondrial genes.
MOLECULAR BASIS OF MUTATIONS
As discussed before, the mechanisms that maintain DNA sequences are remarkably precise, but they are not perfect. With a few exceptions, cells do not have specialized mechanisms for creating changes in the structures of their genomes: changes depend instead on accidents and mistakes. Errors in DNA replication, DNA recombination, or DNA repair can lead to either simple changes in DNA sequence or to largescale genome rearrangements.
In medical genetics, mutation is the process whereby genes change from one allelic form to another. Mutations can lead to loss of function or to a new function of the gene. Genes mutate randomly, at any time and in any cell of an organism. Mutations in germ-line cells can be transmitted to progeny, but somatic mutations cannot. Mutations can be passed from generation to generation, following mendelian inheritance, or they can represent new mutations (sporadic mutations) that occur in the sperm or egg of the parents or in the embryo.
Geneticists recognize two different levels at which mutation takes place. In gene mutation, an allele of a gene changes, becoming a different allele. Nowadays, point mutations typically refer to alterations of single base pairs of DNA or to a small number of adjacent base pairs. At the other level of hereditary change— chromosomal mutation—segments of chromosomes, whole chromosomes, or even entire sets of chromosomes change. The effects of chromosome mutations are due to the new arrangements of chromosomes and of the genes that they contain.
Single-Gene Mutations.
Point mutations are classified in molecular terms, and these nucleotide substitutions can result in several molecular outcomes. Although point mutations are often considered to occur randomly, there are mutational hot spots in the genome, commonly at CG dinucleotides, and mutations tend to recur at such sites. Transitions, which exchange one pyrimidine for the other or one purine for the other, are more common than transversions, which exchange one pyrimidine for a purine or vice versa. These substitutions can result in several molecular outcomes:
Missense mutations occur when a single nucleotide substitution alters the sense of a codon and a different amino acid is added during protein synthesis.
Nonsense mutations occur when a single nucleotide substitution convert a codon for an amino acid into a termination codon. This change results in the premature termination of translation and a truncated polypeptide.
Promoter mutations alter the transcription of the gene or generate instability of the mRNA, thereby reducing the production of the relevant protein.
mRNA splicing mutations occur in the consensus sequences at the exon-intron boundaries resulting in the adjacent
exon to be spliced out and therefore, a shortened protein chain. In addition, abnormal splicing can also occur when a point mutation create a new or cryptic splice site with complex consequences because splicing may remove part of an exon and include intron sequences.
Frame-shift mutations result from any addition or deletion of base pairs that is not a multiple of 3 changing the reading frame of the DNA resulting in different amino acids from that point on and frequently chain termination.
Finally, it is important to mention single nucleotide polymorphisms (SNPs). These are simply points in the genome sequence where one large fraction of the population has one nucleotide, while the other has another. While most of the SNPs and other common variations in the human genome sequence are thought to have no effect on phenotype, a subset of them must be responsible for nearly all of the heritable aspects of human individuality. A major challenge in human genetics is to learn to recognize those relatively few variations that are functionally important (i.e., contribute to genetic disorders) against the large background of neutral variation that distinguishes the genomes of any two human beings. Genome-wide associations studies (GWAS) looking at millions of SNPs are designed to assess these contributions.
Chromosomal Mutations.
Chromosomal abnormalities are more frequent than all the single disorders together and result from disruptions in the normal number or the structure of the chromosomes (7, 8). An abnormal chromosome number, called aneuploidy, occurs in approximately 4% of pregnancies. Tetraploidy refers to four copies of the full set of chromosomes (for a total of 92), triploidy to three copies (69 chromosomes), trisomy to one chromosome pair having an extra chromosome (47 chromosomes), and monosomy to one chromosome of one pair being absent (45 chromosomes). Finally, unipaternal disomy refers to the concept that individuals have cells that contain two chromosomes of a particular type that have been inherited from only one parent. Isodisomy exists when one chromosome is duplicated, and heterodisomy when both homologs have been inherited form one parent.
Abnormalities in the chromosomal structure occur less frequently than numerical abnormalities. These abnormalities are called balanced if the chromosome set has the normal complement of DNA and unbalanced if there is additional or missing DNA. Balanced rearrangements do not usually have a phenotypic effect because all of the genetic information is present, but arranged differently. However, these rearrangements can disrupt a gene at the site of the break resulting in that specific gene dysfunction. Unbalanced rearrangements alter the normal amount of genetic information and commonly result in an abnormal phenotype. The basic molecular mechanisms resulting in structural chromosomal abnormalities include deletion (a section of the chromosome is absent); duplication (an extra section of the chromosome is present); translocation (a portion of a chromosome is exchanged with a portion of another chromosome); and inversion (a broken portion of a chromosome reattaches to the same chromosome in the same location, but in a reverse direction).
PATTERNS OF INHERITANCE IN MEDICAL GENETICS
In the study of genetic disorders, four mendelian patterns of inheritance are distinguishable by pedigree analysis: autosomal dominant, autosomal recessive, X-linked dominant, and X-linked recessive (9, 10). In addition, there are several other forms of inheritance that do not follow the inheritance laws of Mendel. These include mutations in mitochondrial DNA, trinucleotide repeats, mosaicism, genomic imprinting, and uniparental disomy.
Autosomal Disorders
Autosomal Dominant.
In these conditions, the gene mutation is located in one of the autosomal chromosomes. The affected phenotype of an autosomal dominant disorder is determined by a dominant allele, that is, the mutation in one of the alleles is sufficient to express the phenotype. In pedigree analysis, the main clues are that the phenotype tends to appear in every generation and that affected fathers and mothers transmit the phenotype to both sons and daughters. This equal representation of both sexes among the affected individuals rules out X-linked inheritance. People bearing one copy of the A allele (A/a) are much more common than those bearing two copies (A/A) (which could be lethal in some cases), so most affected individuals are heterozygous. The homozygotes are usually much more severely affected than heterozygotes, often resulting in perinatal death (Fig. 2-4).
These disorders appear in every generation because the abnormal allele of the affected individual must have come from one of the parents in the preceding generation. However, abnormal alleles can arise de novo by the process of spontaneous mutation, so this fact has to be kept in mind when analyzing the type of inheritance in that family and for genetic counseling.
Many autosomal dominant disorders have major musculoskeletal anomalies. These disorders include many chondrodysplasias, osteogenesis imperfecta, Marfan syndrome, Ehlers-Danlos syndrome, Charcot-Marie-Tooth disease types IA and IB, and neurofibromatosis 1.
Autosomal Recessive.
The affected phenotype of an autosomal recessive disorder is determined by a recessive allele, and the corresponding unaffected phenotype is determined by a dominant allele. For the phenotype to be expressed, the individual has to have both alleles mutated (a/a). The presence of an A allele is enough for not expressing the phenotype. The two key points for an autosomal recessive disorder are that (a) generally the disease appears in the progeny of unaffected parents and (b) the affected progeny include both males and females. When we know that both male and female children are affected, we can assume that we are dealing with simple mendelian inheritance, not sex-linked inheritance.
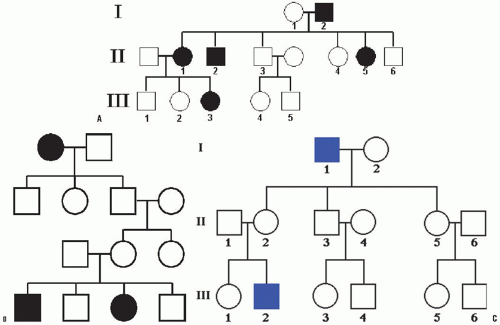 FIGURE 2-4. Pedigree of an autosomal dominant disorder. A: Note affected individuals in every generation and both males and females transmit the disorder to both sons and daughters. B: Note the disorder appears in the progeny of unaffected parents and the affected progeny include both males and females. C: Note that none of the male offspring of an affected male are affected, nor will they pass the condition to their offspring (lack of male-to-male transmission). This is due to the fact that a son obtains his Y chromosome from his father, so he cannot normally inherit the father’s X chromosome. On the other hand, all the daughters of an affected male are “carriers” bearing the recessive allele masked in the heterozygous condition (X/x), so half of their sons will be affected. |
The pedigrees of autosomal recessive disorders tend to look rather bare, with few black symbols representing affected individuals. In general, a recessive condition shows up in groups of affected siblings, and the people in earlier and later generations tend not to be affected. This is due to the fact that if the condition is rare, most people do not carry the abnormal allele, and those who do are heterozygous (A/a for example) for it rather than homozygous (A/A). The basic reason that heterozygous are much more common than homozygous is that, to be recessive homozygote, both parents must have had the a allele, but, to be heterozygote, only one parent must carry the a allele. The clinically normal parents are called carriers. Carrier frequency varies considerably but, for common autosomal recessive disorders, it is approximately 1 in 45 individuals. Autosomal recessive traits are more frequent in consanguineous marriages, particularly if the mutant gene is rare.
X-Linked Disorders.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


