6 General affections of the skeleton
A large number of general affections of the skeleton have been described. Even an incomplete description could occupy a large volume. Fortunately many of these affections are so rare that it is unnecessary for the student to concern himself with them. Most of the others require only brief consideration. Clearly, many of the affections to be described have a congenital basis and many of them cause deformity; so there is inevitably some overlap between this chapter and Chapter 5.
CLASSIFICATION
The following classification is based on that of Wynne-Davies and Fairbank (1976).
BONE DYSPLASIAS
ACHONDROPLASIA
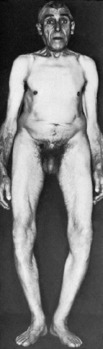
Clinical features. Achondroplasia is apparent at birth, the child being strikingly dwarfed, with very short limbs that are out of proportion to the trunk: shortness is especially marked in the proximal segments of the limbs (Fig. 6.1). Adult achondroplasts are seldom more than 130 cm (4 feet 3 inches) in height. The hands are short and broad, the central three digits being divergent and of almost equal length (‘trident’ hand). The head is slightly larger than normal, with a bulging forehead and depressed nasal bridge. There is marked lumbar lordosis, often with thoracic kyphosis, which may occasionally lead to compression of the spinal cord. There is no mental impairment and life expectancy is usually normal.
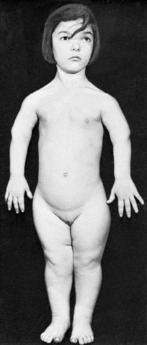
OSTEOGENESIS IMPERFECTA (Fragilitas ossium)
Osteogenesis imperfecta is a congenital and inheritable disorder – or more probably a heterogeneous group of disorders – in which the bones are abnormally soft and brittle, on account of defective collagen formation. In addition to the bones, other collagen-containing tissues such as teeth, skin, tendons, and ligaments may be abnormal. It is usually transmitted as an autosomal dominant, but in a severe variant of the disease the parents are normal and a fresh gene mutation or autosomal recessive inheritance is postulated.
Clinical features. In the worst cases, which occur sporadically rather than from inheritance and probably represent a distinct entity, the child is born with multiple fractures and does not survive. In the less severe examples fractures occur after birth, often from trivial violence. As many as fifty or more may be sustained in the first few years of life. The fractures unite readily, but in the more severe cases marked deformity often develops, either from malunion or from bending of the soft bones (Fig. 6.2), and such patients may be badly crippled. In the milder cases there is a tendency for fractures to occur less frequently in later life.
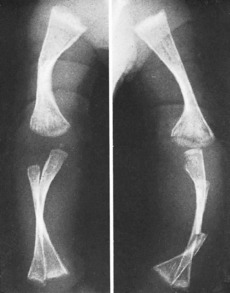
MULTIPLE HEREDITARY EXOSTOSES (Diaphyseal aclasis)
Pathology. The fault is in the epiphysial cartilage plate. Nests of cartilage cells become displaced and give rise to bony outgrowths, which are capped by proliferating cartilage. Growth of the exostoses may cease at skeletal maturity. These exostoses, or osteochondromata, constitute one type of benign bone tumour (p. 109). The number of outgrowths varies; often there are between ten and twenty, of varying size. In severe cases the process of remodelling by which a bone attains its normal adult shape is impaired, and there may be marked deformity, with reduction of longitudinal growth and consequent shortness of stature. Malignant change in the cartilaginous cap of one of the tumours can lead to the development of a chondrosarcoma (the lifetime risk is approximately 5%).
Radiographs show the bony outgrowths. In severe cases the bones are broad and ill modelled (Fig. 6.3).
MULTIPLE ENCHONDROMATOSIS (Dyschondroplasia; Ollier’s disease1)
In dyschondroplasia masses of unossified cartilage persist within the metaphyses of certain long bones, and the growth of the bone is retarded. The condition becomes evident in childhood, but the cause is unknown. In contrast with multiple hereditary exostoses, just described, heredity plays no part.
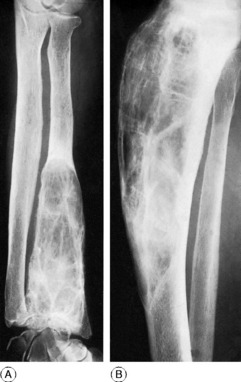
Any bone formed in cartilage may be affected. The more rapidly growing ends of the femur and tibia (that is, the ends near the knee) and the small long bones of the hands and feet are particularly common sites. There is a tendency for the disorder to affect one side of the body predominantly, or even exclusively. When a major long bone is affected interference with growth at the epiphysial cartilage adjacent to the lesion may lead to serious shortening and distortion of the bone (Fig. 6.4A). When skeletal growth ceases the masses of cartilage may ossify. Rarely one of the tumours undergoes malignant change, to become a chondrosarcoma (p. 117).
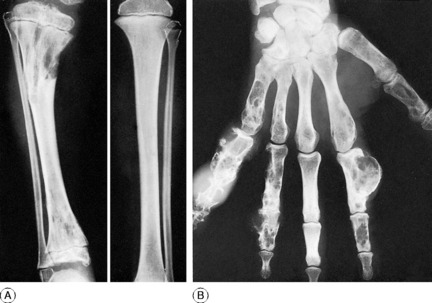
Clinical and radiographic features. A limb affected by dyschondroplasia is usually short and may be markedly deformed. The hands may be grotesquely enlarged by multiple cartilaginous swellings or outgrowths. Radiographs show multiple areas of transradiance in the affected bones (Fig. 6.4).
Treatment. Osteotomy may be required to correct deformities resulting from uneven growth of bone. If there is marked discrepancy in the length of the lower limbs a leg equalisation procedure (p. 46) may be advisable. Lesions in the hand may be curetted and packed with bone chips.
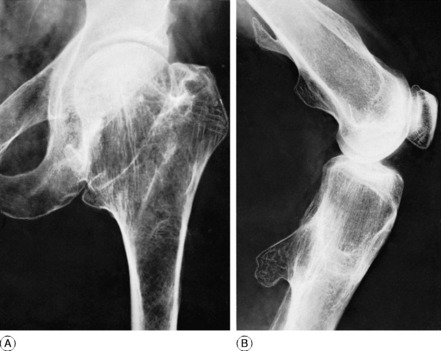
PAGET’S DISEASE1 (Osteitis deformans)
Clinical features. The affection seldom begins before the age of 40, but a childhood form is recognised. Often there are no symptoms, the condition being discovered incidentally during routine radiographic examination. When long bones are affected pain is sometimes complained of, but it is by no means an invariable feature. Bending of the softened long bones leads to deformity, usually in the form of anterior and lateral bowing of the femur or tibia (Fig. 6.5). Thickening may be obvious clinically, especially in the case of the tibia or the skull. Thus if the patient wears a hat he may notice that he requires progressively larger sizes. More importantly, bone thickening may also cause constriction of foramina in the skull, with risk of damage to a nerve – for instance the optic nerve or the auditory nerve.
Imaging. The main radiographic features (Figs 6.6 and 6.7) are:
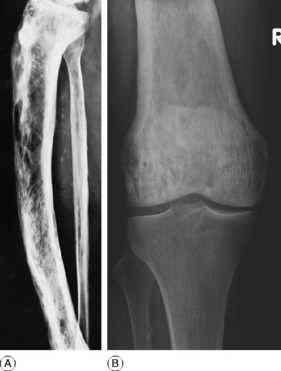
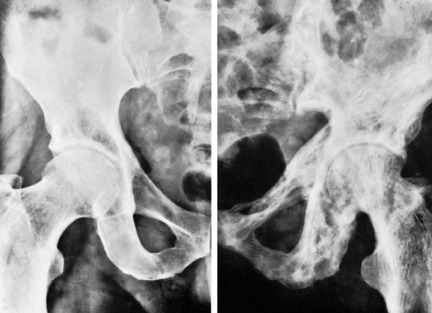
The long bones are often shown to be bowed, the pelvis may be deformed, the vertebrae may be compressed and the vault of the skull thickened. Radioisotope scanning with 99mtechnetium shows markedly increased uptake in the affected bones.
Both drugs inhibit bone resorption. They reduce bone turnover, with lowering of plasma alkaline phosphatase, and both are effective in relieving bone pain in a high proportion of cases. Treatment must, however, be prolonged. Unlike calcitonin, which must be injected (though nasal spray and suppository forms are being tried), a bisphosphonate compound may be taken by mouth. Moreover its benefit often persists for up to six months after the drug has been discontinued; so it is the preferred drug in the first instance, on the basis of cost and ease of administration.
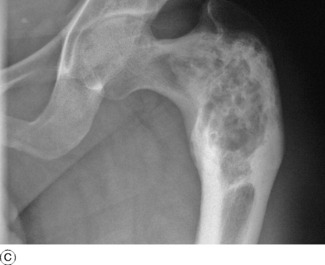
POLYOSTOTIC FIBROUS DYSPLASIA
Replacement of bone by fibrous tissue forms a conspicuous part of several unrelated bone diseases. In two conditions in particular, fibrous replacement is the predominant change. In one of these – parathyroid osteodystrophy – the changes are associated with hyperparathyroidism (p. 73). In the other, now to be described, there is fibrous replacement without any evidence of excessive parathyroid secretion.
Radiographs of the affected bones show well-defined transradiant areas which often have a characteristic homogeneous or ‘ground-glass’ appearance: these changes may be localised and patchy rather than uniform. The lesions are in the shaft and metaphyses rather than the epiphyses. When the lesion is extensive the cortex is expanded and thin, and the bone is bent when weight-bearing, producing deformities such as the ‘shepherd’s crook’ in the proximal femur (Fig. 6.8). Sometimes the lesion has a honeycombed appearance.





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


