Chapter 1 Epidemiology of Systemic Lupus Erythematosus
INTRODUCTION
Systemic lupus erythematosus (SLE) is one of the most common autoimmune diseases.1 It often associates with severe morbidity; mortality rates higher than those of the general population are well recognized.2 Understanding the distribution of SLE across different populations may help estimate the burden that it imposes at individual and societal levels. Given that SLE affects mainly individuals during their adult years,3–9 it has the potential to account for years of loss productivity10–12 as well as for functional losses affecting the same, and therefore, the quality of these patients’ lives.13–17
During the past few decades different SLE studies5,8,9,18–24 have provided valuable information about the distribution of SLE, and its course and outcome. While some studies have addressed the impact certain nonmodifiable factors such as age at disease presentation, gender and ethnicity may exert, others have addressed the role that socioeconomic factors have in outcomes such as damage accrual and mortality. In the following, we address lupus worldwide, the impact of age at disease onset, the impact of gender, the impact of ethnicity, and mortality trends.
Lupus Worldwide
SLE has been recognized in all five continents, although it appears to be more common in Europe, the Americas, and Asia than in Australia25 and Africa.26 Of interest, in individuals of African ancestry, the disease appears to be quite rare in Africa but common in individuals of African ancestry living the United States, the Caribbean Islands, the United Kingdom, and Continental Europe.
Analyses of the population burden imposed by SLE are hampered by the different sampling and recruitment methodologies used in the studies reporting prevalence and incidence rates. Several issues need to be addressed to better understand the differences in the rates reported. First, a case definition should be included in the report. Studies conducted prior to the establishment of classification criteria by the American Rheumatism Association (ARA), now the American College of Rheumatology27 (revised in 198228 and modified in 199729), used various different case definitions; it is more than likely these studies could not have captured cases of mild SLE. The ACR criteria are now widely used in the clinical setting despite the fact that they were intended for the research setting; moreover, they were validated using prevalent rather than incident cases of SLE, because some time may elapse between the first disease manifestation and the accrual of four ACR criteria, which is a requirement for a patient to be classified as having SLE.30
Second, the method used to gather data may yield different rates, as they have variable case-capture sensitivity and specificity.31,32 Patient questionnaires,31,33–35 self-reported physician diagnosis,36–38 medical records review,39 or multiple assessment methods with capture-recapture techniques2 have been used.
Table 1.1 depicts SLE incidence rates reported over the last three decades. Although difficult to compare as already noted, it seems that there is a trend toward an increase in the incidence of SLE2,40; if this increment is real, or if it simply reflects a more accurate case ascertainment or the inclusion of milder cases is difficult to determine.
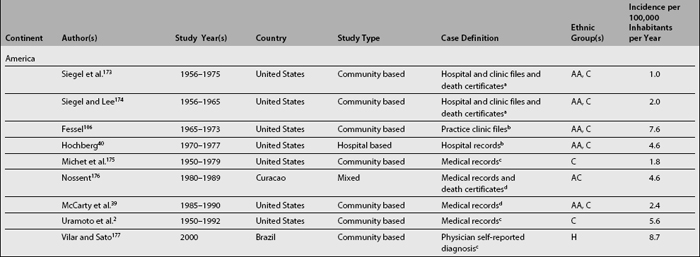
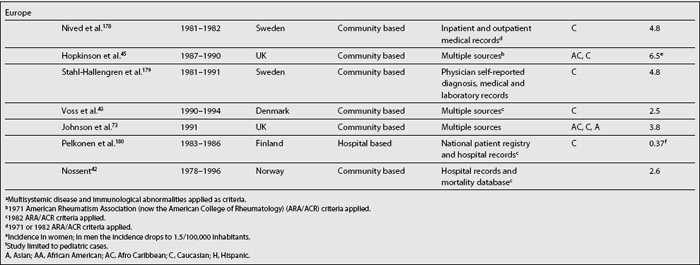
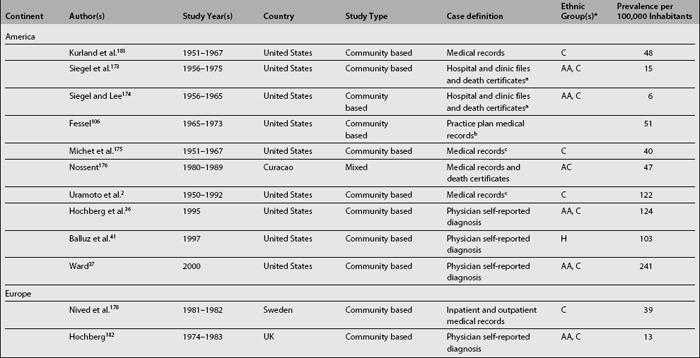
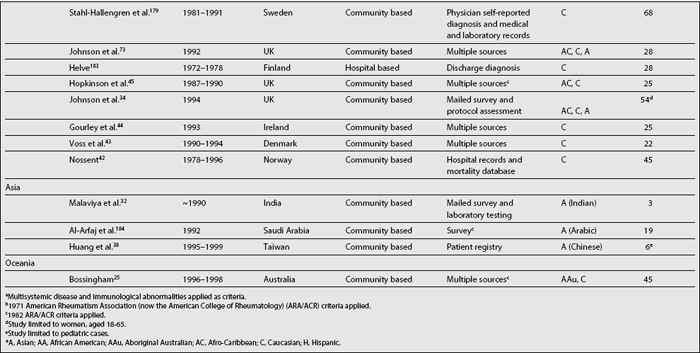
As shown in Table 1.2, SLE prevalence rates vary widely around the world; higher rates have been reported in the United States2,36,37,41 than in countries in Europe,34,42–45 Asia,32,38 and Oceania.25 There is also a tendency toward higher prevalence rates now than in the past.2,46 Possible reasons underlying this phenomenon are the inclusion of milder cases, improved case ascertainment and capture methods, and finally, the accrual of cases over time as a result of improved survival rates and consequently of patients experiencing a longer disease duration.46,47
Impact of Age at Disease Onset
Pediatric lupus, that is SLE with onset or diagnosis before age 16 years, accounts for approximately 8% to 15% of all SLE cases48–51; yet SLE rarely occurs before age 5 years, being more common after age 10.52–55 The female-to-male ratio seems to be lower than among the adult-onset lupus (6:1), yet a female-to-male ratio closer to that of the adult-onset lupus population ranging from 7 to 18:1 has been reported in some studies from Western and Middle Eastern countries.51,54,55 However, gender does not seem to impose a different prognosis in pediatric SLE.53 Lupus in the pediatric age group usually presents with severe disease manifestations including a high proportion of patients with major organ system involvement; this is particularly the case for renal51,54,56,57 and neuropsychiatric involvement.57–60 Not surprisingly, therefore, the domains of the damage index most frequently affected are the renal and neuropsychiatric domains.52 Few studies have addressed survival in pediatric-onset lupus; in the few studies addressing this, survival rates as high as 80% at 10 years have been reported.60,61
Once thought to be a rare condition, late-onset lupus (generally considered if onset occurs at age 50 and later),62–65 is being increasingly recognized beyond the sixth decade of life. According to a meta-analysis conducted in the late 1980s that included nine studies, late-onset lupus comprised up to 18% of all patients.66 Given that life expectancy has increased significantly over the last century, and it is expected to continue to increase,67 the increased occurrence of late-onset lupus is not surprising as there are more individuals at risk of developing it. The female-to-male ratio also tends to be lower than among adult early-onset patients with a female-to-male ratio of 2.6 to 5.5:1.63–65,68,69 Late-onset lupus patients tend to have a more insidious onset,62,70 to be less likely to have major organ system involvement,65 and to have lower degrees of disease activity.63 In the meta-analysis mentioned, the authors concluded that these patients have more serositis, interstitial lung disease, Sjögren’s syndrome, and anti-La antibody positivity than their younger counterparts.66 Despite these relatively less-serious disease manifestations, late-onset lupus patients may have a poor outcome, both in terms of morbidity (damage accrual)22,71,71a and mortality.23,50,64,70 The disease itself, plus other age-related factors (comorbidities) probably exert a synergistic negative effect explaining these findings.
Impact of Gender
The most striking gender-related difference in SLE is in its incidence (and prevalence). SLE is much more frequent among women than among men with ratios of 6 to 14:1 reported in the literature.6,39,72 These ratios vary significantly, however, probably as a result of other variables such as ethnicity and age at disease presentation and the relative underascertainment of the disease in men.73 The female preponderance observed probably reflects the role that sex hormones have in the pathogenesis of the disease.74 This relationship has been well demonstrated in murine models of SLE75–78; furthermore, the peak incidence in women occurs during their reproductive years. The disease also tends to flare up during periods of hormonal changes, especially during pregnancy,79–81 or with the use of oral contraceptives and hormone replacement therapy.82–86 The hypoestrogenemic state that occurs during menopause appears, however, not to be protective of disease activity and damage accrual; in fact, it has been suggested that age rather than menopausal status is a strong independent predictor of damage accrual and of vascular events in women with lupus.87 Similarly, the use of hormone replacement therapy appears not to be an independent predictor of disease activity, severe flare-ups, and damage accrual88; however, mild to moderate flare-ups have been reported in the SELENA trial.89
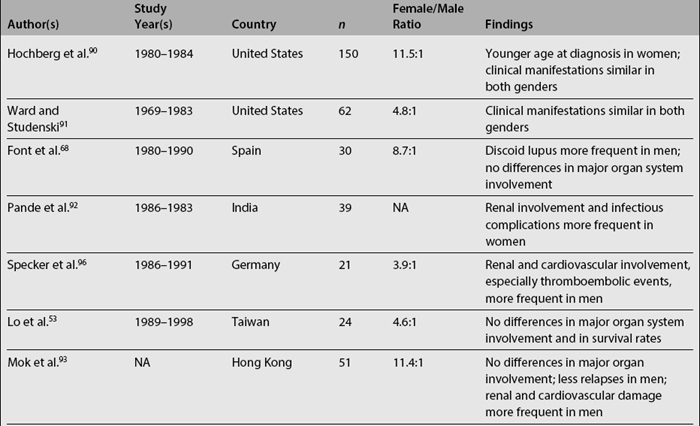
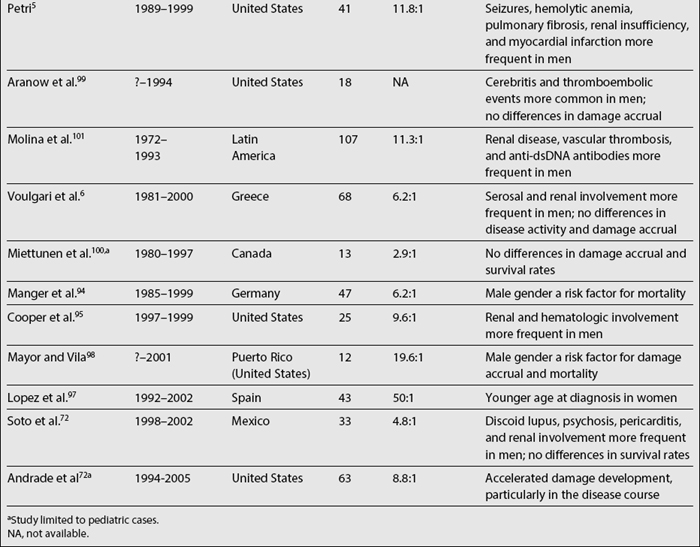
Data on the relationship among gender, clinical manifestations, and disease outcome are somewhat more controversial. Women tend to develop lupus at a younger age than men, while men tend to have serosal and renal involvement more frequently than women. Finally, men tend to accrue more damage and to experience lower survival rates than women, although there is some degree of variability across studies, as noted in Table 1.3.5,6,53,68,72,90–101 It should be emphasized that some of these studies include a relatively small number of patients (20 or less) making their conclusions less reliable.
Impact of Ethnicity
Terms such as race, ethnicity, or ancestry are not interchangeable. Race implies genetic homogeneity, which does not exist in humans; for example, Hispanics in the United States and Asians in England and in the United States include different subgroups or categories of individuals within each group.18,19,102,103 Individuals categorized as Caucasians or white are equally heterogeneous. The Institute of Medicine (United States) has recommended that the term “race” be banned from the scientific literature, and that ethnicity be used instead. This term is a much broader self-defined construct. Ethnic groups are defined on the basis of geographic, social, cultural, and religious characteristics; patients of the same ethnic group have the potential to exhibit a similar genetic background, particularly within ethnic subgroups. Thus, not surprisingly, the variable phenotypic expression of several disorders, SLE among them, among individuals of different ethnic groups has been recognized. This variability cannot be solely explained by genetics-related factors, given the tight association between some socioeconomic indicators of disadvantageous status and defined ethnic groups.
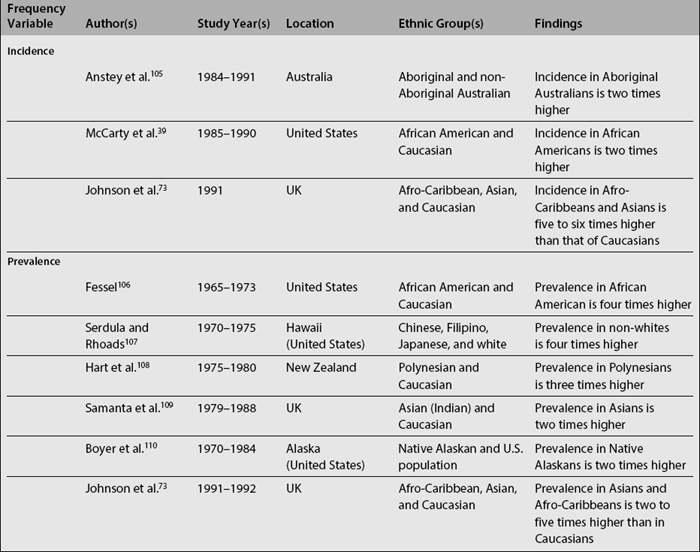
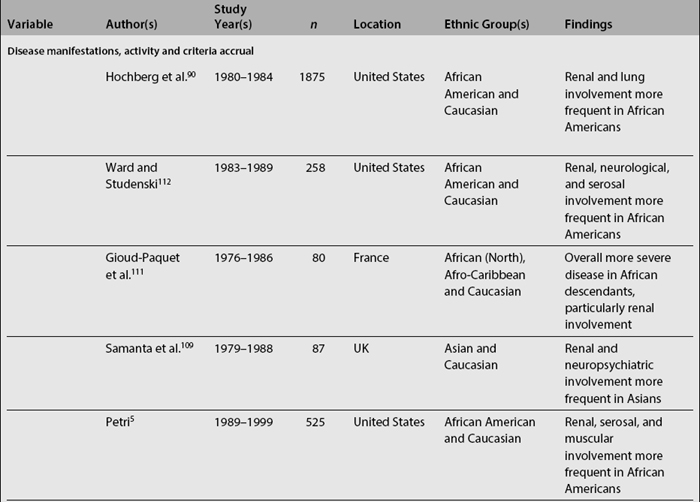
In SLE, differences among ethnic groups can be found in the incidence and prevalence of the disease,39,73,104–110 in its course (disease activity and clinical manifestations)4,5,8,18,90,109,111–114 and in its mediate (damage accrual)22 and long-term (mortality)9,47,60,105,115–141 outcomes.142,143 As noted in Table 1.4, it has been shown that patients from minority populations of African (living in the United States, Caribbean Islands, United Kingdom, or Continental Europe) or Asian ancestry tend to show a higher SLE incidence and prevalence, along with a more severe disease course and outcome.4,5,90,111,112,114,140,144 Similarly, the disease is more frequent among Aboriginal than non-aboriginal Australians.105 These patients as a group tend to have more abrupt disease onset, more severe clinical manifestations, and an overall higher degree of disease activity.145 Hispanics, African Americans, and Asians also tend to have more hematologic, serosal, neurologic, and renal involvement, regardless of age and gender.4,8,18,109,112,113 Patients of non-Caucasian ethnicity also accrue more damage over time22 and faster146 than Caucasians; they also develop specific damage more often (renal and integument)22,147 and exhibit higher mortality rates when compared with Caucasians.23,107,140 These data are summarized in Table 1.5.
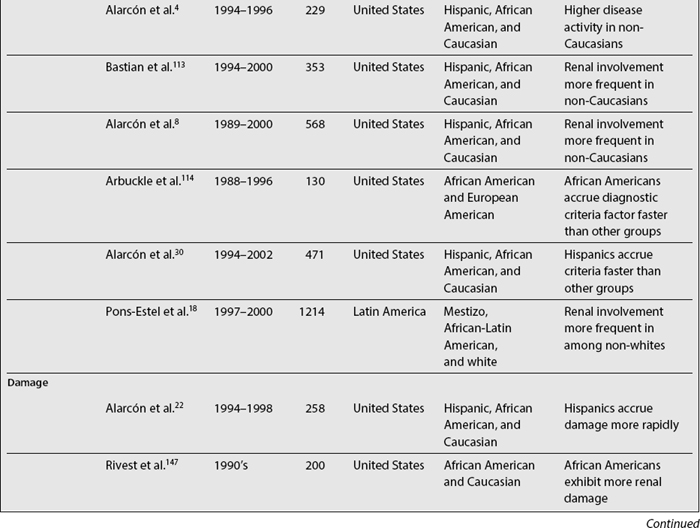
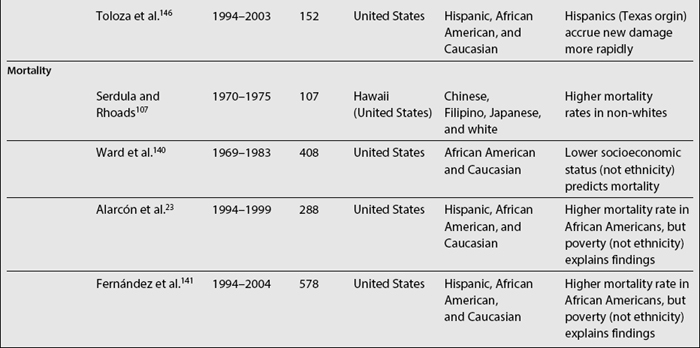
It is very likely that the differences observed among ethnic groups early in the course of the disease reflect the genetic component of ethnicity, whereas the ones observed later in its course reflect nongenetic factors such as those of a socioeconomic-demographic, behavioral, and/or psychological nature.
Mortality Trends
Considered in the first half of the twentieth century a uniformly fatal disorder, patients with lupus are now expected to live years if not decades after diagnosis. Survival analyses were first used in medicine in the early 1950s when Merrell and Shulman115 published their landmark paper demonstrating a survival rate of less than 50% at 5 years (pre-corticosteroid patients). This figure contrasts with those reported over the last two decades showing survival rates at 5 years of 90% and above.94,131,139,148 This improved survival can be explained by earlier diagnosis of the disease, the diagnosis of milder cases (who may not have been diagnosed in years past), the introduction of glucocorticoids and possibly of immunosuppressants and the availability of effective therapeutic interventions for comorbid conditions such as dialysis, antibiotics, and antihypertensive agents. Furthermore, during the same period of time, the improvement in survival in lupus has been greater than the one observed in the general population,149 as evidenced by standardized mortality ratios (SMR) that have declined from 10.1 in the 1970s, to 4.8 in the early 1980s and to 3.3 in the 1990s.150 However, life expectancy in SLE patients is still below that of the general population,2,151,152 which means that efforts need to be directed toward unraveling the pathogenesis of the disease—the factors affecting its course and outcome—particularly those affecting survival.
Table 1.6 summarizes various SLE survival studies. As noted before, the reasons underlying the differences in survival rates between older and more recent publications are probably multiple; however, differences in survival rates still persist even in later reports. The basis for such discrepancies relates mainly to characteristics of the cohort being studied including the time at which patients are recruited into the cohort, the patients’ sociodemographic background, the length of follow-up, and the method used for the analyses. For example, mortality in SLE has been reported to be higher during the first few years of the disease132,152,153; thus, inception cohorts, which include patients who otherwise will be censored in other cohorts, will provide lower survival rates than noninception cohorts. Although in many cases disease onset and disease diagnosis are not the same, it is very hard to clearly establish disease onset in patients unless disease manifestations evolve over a relatively short time period; thus, disease diagnosis is the starting point in most studies to date even though this may artifactually shorten disease duration and affect survival rates. Another issue to consider is the length of follow-up of patients in the cohort as well as the rates of loss to follow-up; the longer the follow-up and the higher the retention rates in the cohort, the more accurate the data will be. The patients’ sociodemographic background is also important when comparing results from different cohorts, given that ethnicity and age, for example, are well-recognized factors influencing survival. Finally, and as noted in Table 1.6, contemporary survival rates in developing countries are comparable to survival rates of years past in developed countries, further emphasizing the importance that socioeconomic factors have in the ultimate outcome of SLE. A very valuable tool to estimate improvement in survival rates is the estimation of such rates within the same cohort over time, avoiding some of the above mentioned problems; for example Urowitz et al.150 compared SMRs in patients from the Toronto cohort at three different time periods. In such analyses, the SMR decreased from 10.1 in the oldest cohort (1970-1977), to 4.8 in the intermediate (1978-1985), and to 3.3 in the most recent (1986-1994).
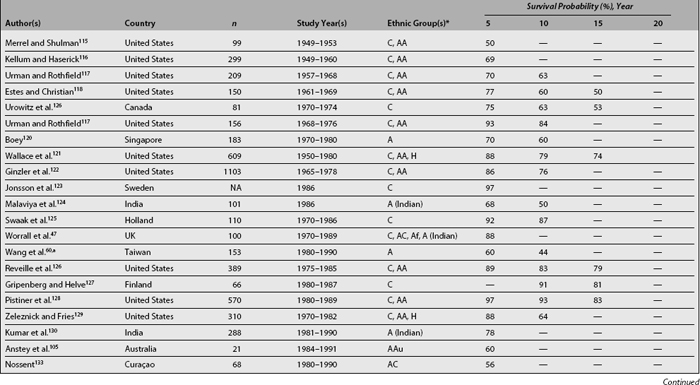
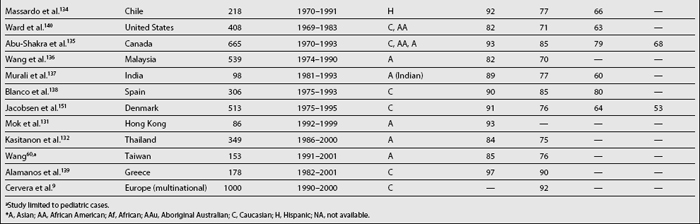
In the 1970s, Urowitz et al. described a bimodal pattern of mortality in lupus119; in that study the authors reported an early mortality peak due mainly to active disease and a later peak due to cardiovascular complications. This bimodal pattern has been later corroborated in other studies; currently, the most important causes of death among SLE patients are still considered to be active disease and infections during the first few years of the disease and complications derived from accelerated atherosclerosis later in the disease course.7,60,135,151,152,154,155
A large body of literature deals with factors predictive of mortality. Variables from the socioeconomic-demographic, clinical, and psychological domains have been implicated as predictors of mortality. Data from various countries have shown that patients of non-Caucasian ethnicity have higher mortality rates, even at younger ages, when compared with Caucasian patients.23,105,107,108,130,140,156,157 Moreover, data from the United States have shown that mortality is not only higher in African American women, but that it is also increasing over time when compared to Caucasian women87,158,159; these data are depicted in Fig. 1.1. Information on other minority groups is scarce, particularly for Hispanics. However, interpretation of the role that ethnicity has in predicting mortality must be done with great caution given that, as already discussed, “ethnicity” could be a proxy for socioeconomic-demographic variables and psychological factors that could be the actual reasons for the differences observed. For example, in the LUMINA (LUpus in MInorities, NAture versus Nurture) cohort, African-American patients exhibited a lower probability of survival in univariable analyses. However, in multivariable analyses, poverty rather than ethnicity has been consistently found to be an independent predictor of mortality; this was the case in analyses performed in a prevalent and relatively young cohort,23 but has been corroborated recently.141 The impact of other socioeconomic variables has also been underscored in other studies; for example, fewer years of formal education have been found to be associated with higher mortality rates in Caucasian patients160 and the lack of health insurance has been found to independently predict mortality in the GLADEL (for Grupo Latino Americano de Estudio de Lupus or Latino American Group for the Study of Lupus) study, a multinational Latin American cohort.18 Ward et al.140 have also found several indicators of socioeconomic status, such as income and type of medical insurance, to be associated with mortality.
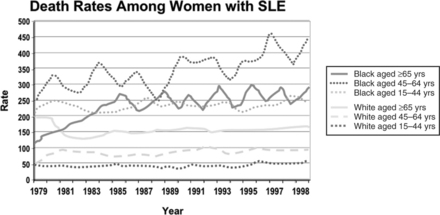
Fig. 1.1 Death rates (x100,000 inhabitants) among women with systemic lupus erythematosus by ethnic group.
(From Sacks JJ, Helmick CG, Langmaid G, Sniezek JE. MMWR Morb Mortal Wkly Rep 2002;51:371-4.)

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


