Diseases of the Hematopoietic System
Michael T. Busch
Amy L. Dunn
Diseases of the hematopoietic system can profoundly affect musculoskeletal form and function. The hematopoietic system consists of the cellular elements in circulating blood, bone marrow, spleen, lymph nodes, and the reticuloendothelial system. This chapter discusses the diseases of the hematopoietic system that have musculoskeletal features that a pediatric orthopaedist would be expected to diagnose and treat. Such diseases can be divided into (a) disorders of the bone marrow, where most of the cells of this system are produced; (b) disorders of erythrocytes/hemoglobin, predominantly involving abnormalities in hemoglobin synthesis or erythrocyte production; (c) disorders of neutrophils and lymphocytes, with accompanying immune deficiencies; (d) disorders of monocytes and macrophages, with abnormalities of metabolism and proliferation; (e) disorders of hemostasis, causing abnormal bleeding or thrombosis; and (f) hematologic malignancies. The orthopaedic evaluation and management are discussed for each disorder, along with recent advances in pathophysiology, molecular genetics, treatment, and prognosis.
BONE MARROW FAILURE DISORDERS
The cellular elements of the hematopoietic system are produced in the bone marrow. Bone marrow failure is characterized by deficient production of one or more cell lines in the bone marrow. Disorders characterized by bone marrow failure can cause anemia, thrombocytopenia, leukopenia, or pancytopenia, depending on which hematopoietic precursors are affected and at what stage of stem cell differentiation the abnormality occurs. There are many bone marrow failure disorders with musculoskeletal manifestations, the most common of which include Fanconi anemia (FA), thrombocytopenia with absent radius (TAR) syndrome, Diamond-Blackfan anemia (DBA), Schwachman-Diamond syndrome (SDS), and cartilage-hair hypoplasia (CHH). The orthopaedist should be familiar with these disorders, because the musculoskeletal manifestations that can cause significant functional disabilities and cosmetic problems may be their first clinically apparent signs.
Fanconi Anemia.
FA is an autosomal recessive disorder characterized by bone marrow failure, physical anomalies, and predisposition to malignancies. The disorder is uncommon, with an incidence of <1 per 100,000 live births (1). The disorder has a slight male predominance 1.3:1(2) and shows no clear racial dependence (3). Multiple genes have been implicated in FA, the most common of which FANCA is located on chromosome 16q24.3 (4). While the exact function of FA proteins is unclear, they likely lead to defects in apoptosis of hematopoietic progenitors (5). Patients with FA show increased chromosomal breakage especially when exposed to DNA crosslinking agents. As skeletal anomalies are the most obvious clinical manifestations of FA apparent at birth, the orthopaedist may be the first physician consulted. Nearly 60% of patients exhibit short stature while nearly 50% manifest upper limb anomalies. The hand and forearm are the sites most often affected, with a variety of radial ray differences (6, 7 and 8). Thumb hypoplasia or absence is common, although thumb duplication and triphalangeal thumb may also be seen. The radius is typically either hypoplastic or absent. It is important to distinguish FA from TAR syndrome. In FA, if the radius is affected, the thumb is also abnormal, while in TAR, if the radius is absent, the thumb is always present. Although radial ray differences are a common manifestation of FA, they are not pathognomonic (9). Nonetheless, any child with a radial ray deficiency should be referred to genetic specialists and/or hematologists for evaluation of the possibility of an underlying hematologic problem. Because 20% of patients with FA have no clinically apparent anomalies at birth, the diagnosis may be delayed.
Other skeletal anomalies such as congenital hip dislocation, spina bifida, and scoliosis have been reported in patients with FA, but not often enough to be considered an integral feature of the disorder. Patients tend to have a variety of facial
abnormalities including micrognathia, epicanthal folds, and a broad nasal bridge. Skin pigmentation anomalies, including café-au-lait spots, are common. As the child grows, growth retardation becomes apparent in approximately 80% of patients (6), often associated with endocrinopathies (10). Renal anomalies are present in approximately one-third of patients with FA, although the exact prevalence is likely underestimated because not all patients in large series have undergone renal diagnostic imaging (11). Cardiovascular and gastrointestinal anomalies can be found in 15% to 30% of patients. The central nervous system is often affected, with microcephaly, hearing loss, eye anomalies, and mental retardation apparent in up to 37% of patients (6).
abnormalities including micrognathia, epicanthal folds, and a broad nasal bridge. Skin pigmentation anomalies, including café-au-lait spots, are common. As the child grows, growth retardation becomes apparent in approximately 80% of patients (6), often associated with endocrinopathies (10). Renal anomalies are present in approximately one-third of patients with FA, although the exact prevalence is likely underestimated because not all patients in large series have undergone renal diagnostic imaging (11). Cardiovascular and gastrointestinal anomalies can be found in 15% to 30% of patients. The central nervous system is often affected, with microcephaly, hearing loss, eye anomalies, and mental retardation apparent in up to 37% of patients (6).
Bone marrow failure occurs in 90% of patients and while it may begin with one cell line often progresses to profound decreases in all cell lines (1). The pancytopenia typical of FA does not typically appear until age 7 or 8 years (12), although it can occur at any age (1). Because patients with FA have increased susceptibility to DNA breakage, the incidence of solid tumors and leukemia is increased in patients with FA. One-fourth of patients with FA have at least one malignancy, and in one-fourth of these patients, the finding of malignancy preceded the diagnosis of FA. The treatment of the upper extremity anomalies is covered elsewhere in this text.
Thrombocytopenia with Absent Radius Syndrome.
TAR syndrome is a rare autosomal recessive disorder characterized by marked thrombocytopenia and absent radii. The radial absence is complete and almost always bilateral (13). The radial clubhands seen in TAR syndrome differ from those in FA in that the thumbs are present in TAR syndrome but are absent in FA if the radii are absent (Fig. 10-1). (See Table 10-1 for a comparison of FA and TAR syndrome.) The thumbs are hypoplastic in about half of patients with TAR syndrome. Thumb and finger function is impaired to varying degrees, and wrist function is abnormal owing to the lack of radius and abnormal carpal bones and musculature. Typically, the ulna is hypoplastic and bowed, as in other causes of radial clubhand. In approximately 40% of the patients, the humerus is hypoplastic or absent, and the shoulder girdle is abnormal in one-third of the patients (14). Major intercalary transverse deficiencies may exist, with hands arising directly from the shoulder girdle (14, 15).
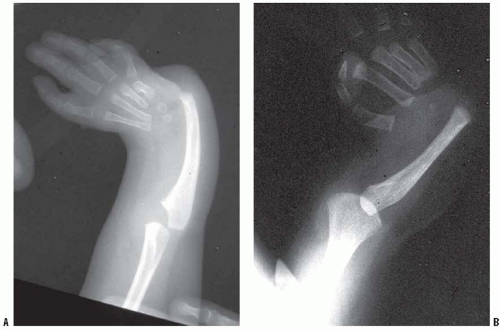 FIGURE 10-1. Forearm and hand radiographs in (A) Fanconi anemia and (B) TAR syndrome. Note the absence of a thumb in the child with Fanconi anemia, as is the case when the radius is completely absent. However, note the presence of the thumb in the child with TAR syndrome, despite complete absence of the radius. |
Lower extremity deformities are also common in TAR syndrome, despite the name of the disorder. In the original description of 40 patients by Hall et al. (14), 40% had lower-extremity deformities. More recent series have estimated an 80% prevalence of lower extremity deformities (16, 17). The severity of upper extremity involvement seems to correlate with the presence and severity of lower extremity involvement (15). As with the fingers, all five toes are typically preserved, even with fibular hemimelia or other lower extremity deformities (15). Knee abnormalities have been studied in detail in 21 patients by Schoenecker et al. (16). Genu varum was present in 18 patients and was apparent at birth in 12 of them. The genu
varum was usually associated with varus laxity of the knee joint rather than a fixed bony deformity. Intra-articular pathology was found to be extensive at surgery in six patients, including concave medial femoral condyles. Internal rotation of the tibia was also common. Patellar anomalies included hypoplasia, instability, and total absence. The deformities tended to progress as the children grew, requiring bracing. Lower extremity deformities usually recurred following corrective osteotomies, although some have reported success in correcting a fixed knee deformity with osteotomy (18). Anomalies of other systems, including cardiac, neurologic, and genitourinary, are reported in one-third of patients (14).
varum was usually associated with varus laxity of the knee joint rather than a fixed bony deformity. Intra-articular pathology was found to be extensive at surgery in six patients, including concave medial femoral condyles. Internal rotation of the tibia was also common. Patellar anomalies included hypoplasia, instability, and total absence. The deformities tended to progress as the children grew, requiring bracing. Lower extremity deformities usually recurred following corrective osteotomies, although some have reported success in correcting a fixed knee deformity with osteotomy (18). Anomalies of other systems, including cardiac, neurologic, and genitourinary, are reported in one-third of patients (14).
TABLE 10-1 Clinical Features of Fanconi Anemia and TAR Syndrome | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
The hematologic manifestation of TAR syndrome is a profound thrombocytopenia that can cause serious bleeding within the first few months of life (14). Bone marrow examination typically reveals infrequent or absent megakaryocytes, but the cause of this is unknown. Viral illnesses can exacerbate the thrombocytopenia, and patients should be kept relatively isolated in the early months to avoid undue viral challenges. Bone marrow transplant is rarely indicated, as most patients resume platelet production spontaneously (19). Elective surgery should be avoided in the first year of life if thrombocytopenia is present. The radial clubhands should initially be splinted and reconstructive procedures performed later.
Diamond-Blackfan Anemia.
DBA is a rare congenital hypoplastic anemia affecting only the erythropoietic cell line and associated with hand and other skeletal anomalies. Approximately 4 in 1 million live births are affected by DBA (20). Typically, the disorder occurs sporadically, but up to 45% of cases are familial. The first identified DBA gene (ribosomal protein RPS19) is located at 19q13. and is thought to have a role in ribosome biogenesis 2 (12, 21, 22). Approximately 30% of patients have associated skeletal anomalies (12). Among upper limb differences, triphalangeal or hypoplastic thumbs are the most common features (12). Radial hypoplasia is uncommon in DBA (23). Patients with DBA also have urogenital anomalies, and cardiac anomalies such as atrial or ventricular septal defects.
Whereas the skeletal anomalies are apparent at birth, the signs of severe anemia do not usually develop until later in infancy. The anemia is typically normochromic and macrocytic, with most patients being identified before 1 year of age. The exact cause of DBA is unknown, and the features of the disorder are highly variable. Bone marrow examination reveals an isolated deficiency of erythroid precursors (12). A predisposition to malignancies may exist in patients with DBA, but as no known impairment exists in DNA repair mechanisms, the predisposition is slight compared to that of FA.
Treatment is evolving (24). Corticosteroids remain the mainstay of treatment in most patients; however, side effects of longterm steroid use is common. For those who do not respond to corticosteroids, treatment options include chronic transfusion therapy, hematopoietic growth factors such as erythropoietin, interleukin-3 (IL-3), and stem cell factor, cyclosporin A, or metaclopromide. Bone marrow transplantation from human leukocyte antigen-matched donors has been utilized (25). Gene therapy has met with some clinical success in a small number of patients (26).
Schwachman-Diamond Syndrome.
SDS is an autosomal recessive disorder causing pancreatic insufficiency, bone marrow hypoplasia, metaphyseal dyschondroplasia, and growth retardation. Patients can appear normal at birth, but typically have low birth weights (27). The first signs of the disorder are attributable to malabsorption, including failure to thrive, growth retardation, and steatorrhea. Severe respiratory infections also occur in the first year of life. Few patients have an uneventful neonatal period.
Patients with SDS are referred to the orthopaedist because of skeletal abnormalities contributing to delayed growth and deformity. Metaphyseal chondrodysplasia occurs in approximately 62% of patients, usually in the proximal femur (27). This lesion in the proximal femur can cause coxa vara, coxa magna, pathologic femoral neck fracture, or pseudoarthrosis (27, 28). Other common sites for chondrodysplasia include
the knees, wrists, spine, and ribs (27, 28). Spinal deformity can include kyphosis and scoliosis. Ribs are typically short and anteriorly flared. Long bone bowing is a common finding and can recur following osteotomies. Clinodactyly was reported in almost half of the patients in one series (27).
the knees, wrists, spine, and ribs (27, 28). Spinal deformity can include kyphosis and scoliosis. Ribs are typically short and anteriorly flared. Long bone bowing is a common finding and can recur following osteotomies. Clinodactyly was reported in almost half of the patients in one series (27).
Bone marrow failure in SDS causes neutropenia in 95% to 100%, thrombocytopenia in 66% to 70%, and anemia in 24% to 50% (27, 29). All three cell lines are affected in 25% of patients (19). Myelodysplasia was found in 7 of 21 patients in one series, and 5 of these developed acute myelogenous leukemia (29). Laboratory studies reveal that the pancreatic insufficiency gives rise to enzymatic abnormalities, including the absence of trypsin, lipase, and amylase in the stool. SDS can be differentiated from cystic fibrosis by a normal sweat chloride test. Hepatic, respiratory, and renal dysfunction also occurs. Neurologic development is usually delayed.
Early mortality is usually from infections. Overall, half of the patients with SDS will live to the age of 35 years, but survival is reduced to 24 years for patients with pancytopenia and to 10 years for patients with leukemia. Early treatment includes oral administration of pancreatic enzymes and aggressive antibiotic treatment of infections. The bone marrow failure may respond to growth factors and androgens, although these treatments are only temporarily effective (19). Bone marrow transplantation with reduced intensity conditioning regimens have shown great promise in curing this disorder (30).
Cartilage-Hair Hypoplasia.
CHH is a rare autosomal recessive disorder characterized by disproportionate, shortlimbed dwarfism, thin, sparse hair, and cellular immunodeficiency. The skeletal manifestations of this condition are covered in detail elsewhere in this text.
The hematologic manifestations of CHH include cellular immunodeficiency and anemia. The degree of immunodeficiency varies greatly (31). Recurrent respiratory tract infections may occur in these patients, and serious illness can result from vaccinations with live viruses. The immunodeficiency is usually due to diminished numbers of T lymphocytes. The anemia of CHH is an integral feature, occurring in 73% of patients (32). Anemia may be severe in infancy, but tends to improve with growth (32).
For patients who have no increased susceptibility to infection and have only mild anemia, often no treatment is required. For more severe immunodeficiency with anemia, bone marrow transplantation may be needed. Bone marrow transplantation can correct the immunodeficiency (33, 34) but not the chondrodysplasia (35).
DISORDERS OF HEMOGLOBIN
Erythrocytes in circulating blood carry oxygen to tissues. Hemoglobin carries the oxygen in erythrocytes. Mutations in the genes that encode for protein synthesis can cause abnormal hemoglobin molecules that affect the form and function of erythrocytes. Iron deficiency and chronic inflammation can also diminish production of hemoglobin, resulting in anemia. Disorders in number, form, or function of erythrocytes can cause significant musculoskeletal pathology, and can complicate the treatment of other musculoskeletal conditions.
Sickle Cell Disease.
Sickle cell disease (SCD) is an inherited disorder of hemoglobin synthesis. The protein component of hemoglobin is composed of four globin chains: two α-globin chains and two β-globin chains. Hemoglobin S refers to hemoglobin containing abnormal β-globin produced by a single base change mutation (GAT to GTT) in the sixth codon of exon 1 in the β-globin gene on chromosome 11 (36). As a result, the molecule polymerizes upon (37) deoxygenation, causing distortion or “sickling” of the erythrocytes that contain the abnormal hemoglobin. Hemoglobin C contains β-globin chains with a glutamic acid-to-lysine substitution at the same position.
Four major types of SCD are recognized, according to the genotype of the β-globin gene and the resulting proportion of hemoglobin S. (a) SS disease results from homozygous inheritance of the hemoglobin S mutation. All hemoglobin is hemoglobin S and the sickling is severe. (b) SC disease results from inheritance of one hemoglobin S allele and one hemoglobin C allele. None of the hemoglobin is normal, but the tendency to sickle is tempered by the presence of hemoglobin C. (c) Sβ+ disease results from inheritance of one hemoglobin S allele and an allele with a β-thalassemia mutation that causes slightly reduced β-globin synthesis. Some normal β-globin is produced, and sickling is less severe. (d) Sβ0 disease results from inheritance of one hemoglobin S allele and an allele with a β-thalassemia mutation that causes greatly reduced β-globin synthesis. Very little normal β-globin is produced, leading to a preponderance of hemoglobin S and severe sickling.
Marrow hyperplasia can lead to osteopenia, biconcave vertebrae, and medullary expansion and cortical thinning due to marrow (38) (Fig. 10-2).
Vascular occlusion causes most of the clinical manifestations of SCD. Several factors cause sickle cells to occlude the microvasculature, including abnormal cell shape, cellular dehydration, and increased cellular adhesion to vascular endothelium. Sickling of erythrocytes is thought to confer resistance to infection by Plasmodium falciparum malaria, contributing to the high prevalence in populations where malaria is common. In the United States, SCD affects 1 in 300 to 1 in 600 African Americans (39, 40). Screening facilitates early diagnosis and treatment, which can improve the clinical course (41).
Vaso-occlusive pain events, or pain crises involving the extremities and the back, are common manifestations of SCD (42). Pain crises are commonly associated with infarcts in the humerus, tibia, and femur (43), although infarcts can occur in any bone in the body. Patients presenting with pain crises rarely have striking findings on physical examination. Swelling and decreased range of motion are usually not present. Fever is variably present. Peripheral leukocytosis and erythrocyte morphology on peripheral blood smears have no diagnostic use in a pain crisis (44). Analgesia is the cornerstone of treatment for
a pain crisis. Hydration is an important adjunct to analgesics. Oxygen supplementation has no proven benefit in a patient who is not hypoxic (45).
a pain crisis. Hydration is an important adjunct to analgesics. Oxygen supplementation has no proven benefit in a patient who is not hypoxic (45).
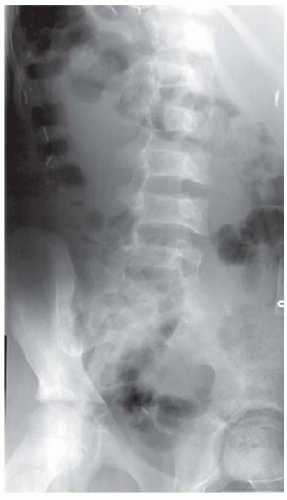 FIGURE 10-2. Oblique radiograph of the spine in a patient with SCD. Note biconcave vertebral bodies. |
Dactylitis, a painful swelling of fingers or toes, occurs in early childhood and is often the first clinical manifestation of SCD and is typically seen in children under the age of 6 years. Onset of dactylitis before the age of 1 year has been suggested to predict a more severe case of SCD (46). The rarity of dactylitis in older children is thought to result from a shift in hematopoiesis from distal sites such as fingers and toes in infants to more central sites in older children (45). Radiographs are initially normal, but may progress to demonstrate periosteal elevation and osteolysis, mimicking osteomyelitis. Cultures of bone aspirates can help in making a diagnosis by differentiating between the two disorders. The pain associated with dactylitis is often mild and is relieved by nonsteroidal anti-inflammatory drugs (NSAIDs) in infants, but can be severe in older children.
Osteomyelitis occurs in patients with SCD and can be difficult to differentiate from a pain crisis. Osteomyelitis is much less common than pain crises; in one study, only 1.6% of patients admitted to the hospital for severe musculoskeletal pain had osteomyelitis (47). Although patients with SCD experience higher rates of Salmonella osteomyelitis than patients without SCD, Staphylococcus aureus is still the most common bacterial pathogen (48, 49 and 50). As microvascular occlusion in the spleen causes repeated splenic infarcts, patients become functionally asplenic and susceptible to infections with encapsulated bacteria such as Streptococcus pneumoniae, Salmonella, and Haemophilus (51, 52 and 53). Intestinal infarcts with translocation of gut bacteria are thought to be responsible for the high rate of infection from Salmonella and other enteric bacteria. Prompt recognition and treatment of osteomyelitis is important. However, it is difficult to differentiate between painful bone infarcts and osteomyelitis, and, therefore, the diagnosis of osteomyelitis is often delayed (54, 55, 56 and 57). The history and physical examination findings are similar in the two conditions. Also laboratory values such as white blood cell count, erythrocyte sedimentation rate, and C-reactive protein are also similar. Imaging is often inconclusive. Plain films are rarely diagnostic. Ultrasound is occasionally effective in detecting subperiosteal fluid collections. Technetium-99m sulfur colloid bone marrow scan followed by technetium-99m methylene diphosphonate bone scan has been reported to differentiate between the two conditions (58), but without proven consistency. Magnetic resonance imaging (MRI) cannot reliably differentiate between sickle infarct and osteomyelitis. The bone marrow manifestations of SCD are primarily related to the hematopoietic marrow hyperplasia, infarction, and perivascular fibrosis. Findings of acute marrow infarction are present in only one-third of cases. Conversely, similar findings on MIR often occur in the absence of clinical symptoms, probably as a result of subclinical (59) infarcts. Gadolinium-enhanced MRI can be useful in distinguishing vascularized inflammatory tissue from abscess, thus guiding aspiration for fluid collection.
The clinical response is important for differentiating between painful crisis and osteomyelitis in patients with SCD. In a painful crisis, symptoms should abate within 24 to 48 hours with hydration and analgesics. If the patient fails to improve, MRI is typically the next step. MRI evidence of intraosseous, subperiosteal, or soft-tissue fluid collection warrants aspiration or surgical drainage.
Septic arthritis is less common than osteomyelitis (57, 60). As opposed to osteomyelitis, septic arthritis is not caused by unusual organisms such as Salmonella (57, 60). The treatment of septic arthritis in patients with SCD follows the principles outlined elsewhere in this text.
Osteonecrosis (ON) of the femoral and the humeral heads is common in patients with SCD (Fig. 10-3). ON of the femoral head is slightly more common than that of the humeral head. The development of ON is related to age and genotype (61, 62). By age 45, nearly one-third of patients have femoral head ON, and nearly one-fourth have humeral head ON. Femoral head ON is bilateral in 54% of the patients, and humeral head ON is bilateral in 67% of the patients. Concomitant femoral and humeral head ON occurs in three-fourths of the patients. The genotype affects the prevalence of ON. As with other manifestations of the disease, patients with SS or Sβ0 disease have a higher incidence of ON than do those with SC or Sβ+ disease (61, 62).
ON may be asymptomatic in the hips and shoulders of children. Abnormalities may show up on radiographs several
years before symptoms appear, and the prognosis is worse in SCD than other causes of femoral head necrosis (61, 63, 64). The age at onset of ON of the femoral head has been reported to correlate with outcome (65), and there may be an impairment of the fibrinolytic pathway in some individuals predisposing them to a worse outcome (66). Plain radiographs and MRI are used for evaluating ON. MRI can delineate the extent and stages of involvement (67, 68). Improving the prognosis and outcomes of ON of the femoral head in patients with SCD is difficult, so the treatment is controversial (69, 70). It roughly parallels that in patients without SCD, as covered elsewhere in this text. Containment and physical therapy may be sufficient in young children with limited involvement of the femoral head. Non-weight-bearing therapy, core decompression, femoral osteotomies, total joint replacement all become options in older and more severely involved hips (71, 72, 73, 74, 75 and 76).
years before symptoms appear, and the prognosis is worse in SCD than other causes of femoral head necrosis (61, 63, 64). The age at onset of ON of the femoral head has been reported to correlate with outcome (65), and there may be an impairment of the fibrinolytic pathway in some individuals predisposing them to a worse outcome (66). Plain radiographs and MRI are used for evaluating ON. MRI can delineate the extent and stages of involvement (67, 68). Improving the prognosis and outcomes of ON of the femoral head in patients with SCD is difficult, so the treatment is controversial (69, 70). It roughly parallels that in patients without SCD, as covered elsewhere in this text. Containment and physical therapy may be sufficient in young children with limited involvement of the femoral head. Non-weight-bearing therapy, core decompression, femoral osteotomies, total joint replacement all become options in older and more severely involved hips (71, 72, 73, 74, 75 and 76).
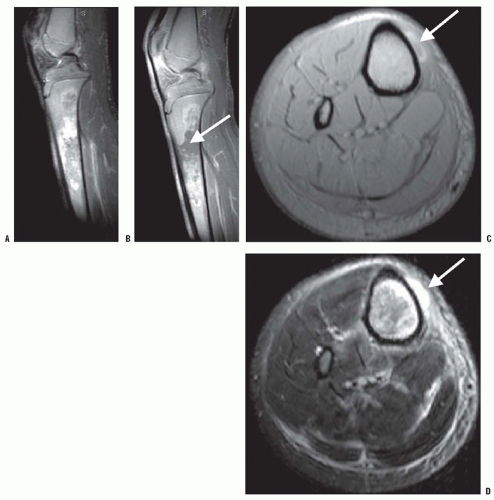 FIGURE 10-3. A 17-year-old boy with SCD presented with symptoms of a painful crisis in his leg. Plain radiographs of his tibia revealed no abnormalities. Failure to respond to hydration after 2 days, along with elevated peripheral white blood cell count and C-reactive protein, prompted investigation with MRI. Sagittal T1-weighted images before (A) and after (B) gadolinium injection demonstrate heterogeneous enhancement throughout a large area of abnormal signal intensity in the marrow of the tibia. An intraosseous fluid collection can be seen (arrow). Axial T1-weighted (C) and T2-weighted (D) images without gadolinium demonstrate an extraosseous fluid collection (arrows) with surrounding edema. Operative corticotomy yielded purulent material. |
Any surgery in patients with SCD should be accompanied by adequate hydration, maintenance of blood volume and oxygenation, and prevention of hypothermia. The use of a tourniquet is allowed, as it does not induce sickling (77). Transfusions are typically given perioperatively to keep the total hemoglobin around 10 g/dL (78).
Pathologic fractures occur in approximately 10% of patients with SCD, usually complicating osteomyelitis (55, 79, 80).
One series (79) found that delayed union, malunion, and joint stiffness complicate 10% to 15% of fractures. However, fractures are not a prominent feature of SCD.
One series (79) found that delayed union, malunion, and joint stiffness complicate 10% to 15% of fractures. However, fractures are not a prominent feature of SCD.
Many other organ systems are affected by SCD. Anemia in SCD is related to erythrocyte fragility and hemolysis. The chronic baseline anemia is generally mild and well tolerated in childhood. However, anemia can be worsened acutely by splenic sequestration, a sudden increase in splenic hemolysis that can be fatal, and by aplastic anemia, a temporary marrow suppression often triggered by parvovirus B19 infection.
Acute chest syndrome (ACS) refers to any new pulmonary infiltrate seen on a chest radiograph in conjunction with fever and chest pain or respiratory symptoms and can be fatal (81). ACS can result from a wide variety of infectious or noninfectious causes, including rib infarcts (82).
SCD also causes genitourinary problems, including enuresis and priapism. Cholelithiasis is common in patients with SCD because of ongoing hemolysis and buildup of bilirubin. Stroke is a common and potentially devastating result of cerebral vasoocclusion or hemorrhage. Infections are a significant risk in infancy and early childhood. Sepsis used to be a major cause of mortality in this age group. The widespread use of penicillin prophylaxis and pneumococcal vaccination in children younger than 5 years reduces the incidence of pneumococcal bacteremia by 84% (83).
Medical treatment of SCD has advanced considerably in recent decades. Hydroxyurea, a chemotherapeutic agent, causes increased formation of hemoglobin F and has been found to reduce the incidence of painful crises and ACS, as well as to reduce the requirement for transfusion in adults (84). Several studies have proven similar efficacy of this drug in children as young as 2 years, although the U.S. Food and Drug Administration (FDA) has not yet approved this drug for use in children. Many other drugs are currently under investigation. Most children now receive pneumococcal vaccine, and it should be highly considered in children with SCD. Bone marrow transplantation has been used in approximately 150 children with severe SCD worldwide, with 92% to 94% survival and 75% to 84% event-free survival (85).
Thalassemia.
The thalassemias are a heterogeneous group of autosomal recessive inherited disorders of hemoglobin synthesis. Together, they represent the most common inherited diseases worldwide (86). The diseases and their treatments can cause an array of alterations in skeletal dynamics that the orthopaedist should be able to recognize.
The many kinds of mutations that are responsible for thalassemia cause deficient or nil production of either α- or β-globin chains. Alpha thalassemia results from mutations in one or more of the four copies of the α-globin gene. One mutation results in a silent carrier state. Mutation of two genes causes a thalassemia trait, characterized by mild normocytic or microcytic anemia. Mutation of three genes causes substantially diminished α-globin production and hemoglobin H disease (named for the stable tetramer formed by the remaining β chains) with moderate hemolytic anemia. Mutation of all four α-globin genes causes hydrops fetalis, which is usually fatal in utero. Beta thalassemia results from mutations in the β-globin gene and is classified as (a) β+ thalassemia, with reduced synthesis of β-globin or (b) β0 thalassemia, with absent β-globin synthesis. An alternate classification of thalassemia is based entirely on clinical severity. Thalassemia major refers to severe disease, thalassemia intermedia refers to moderate disease, and thalassemia minor refers to mild disease.
Among the α thalassemias, hemoglobin H disease is the most often seen clinically. Children generally present with moderately severe anemia, splenomegaly, and cholelithiasis, which may occur in response to oxidative stress caused by infections, fever, or certain medications (87). Patients with thalassemia major (homozygous β thalassemia) develop severe anemia, with hemoglobin in the 3 to 4 g/dL range within the first 6 months of life, as fetal hemoglobin production wanes. Thalassemia major requires frequent transfusions in order to maintain health and prolong life expectancy beyond 5 years of age. Transfusions are generally started when the anemia becomes clinically detrimental and are aimed at keeping hemoglobin levels more than 9.5 to 10.5 g/dL. Thalassemia intermedia typically presents in the second year of life with a less profound anemia (86).
The skeletal manifestations of the thalassemias, which may occur as a result of both the anemia and its treatments, include marrow hyperplasia, short stature, skeletal dysplasia, and osteopenia. Without transfusions to correct the severe anemia in thalassemia major, erythropoietin secretion increases. The resulting marrow hyperplasia causes widening of the medullary cavities and thinning of the cortices of long bones (Fig. 10-4). This process is initially apparent in the hands and feet, where the tubular bones become rectangular and then convex. Premature closure of physes, especially in the proximal humerus, can also occur (88). Marrow hyperplasia can cause dramatic expansion of calvarial bones (89). Marrow hyperplasia in the spine is associated with back pain in adults with thalassemia who started transfusions after 3 years of age (90). Extramedullary hematopoiesis commonly occurs in the liver, spleen, and chest. Extramedullary hematopoiesis in the paravertebral space can cause spinal cord compression (91, 92, 93 and 94). MRI is helpful in detecting and evaluating this process in the spine. Surgical decompression, radiation therapy, and transfusions are treatment options. Marrow hyperplasia from severe anemia is not often seen today, because of the use of maintenance transfusions.
Growth disturbance can result from the effects of transfusion-induced iron overload on the anterior pituitary gland and hypothalamus. Endocrinopathies resulting from iron overload include decreased growth hormone (GH) release or GH resistance (95), delayed puberty and hypogonadism (96), and hypoparathyroidism. In one series of transfusion-dependent patients with thalassemia major (97), 8% of boys aged 7 to 8 years had short stature (less than third percentile), as well as 12% and 15% of older boys and girls, respectively. The short stature tends to be disproportionate, with a relatively short trunk (98). The correction of GH deficiency and the
induction of puberty with gonadotropins partially correct this growth disturbance (96, 98).
induction of puberty with gonadotropins partially correct this growth disturbance (96, 98).
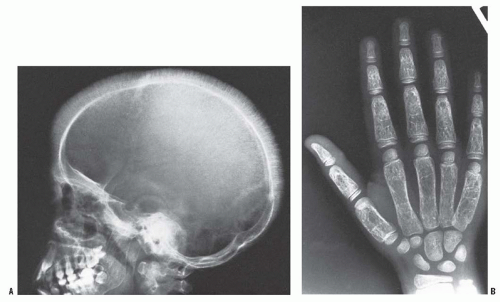 FIGURE 10-4. A: Lateral radiograph of the skull in a 11-year-old boy with thalassemia major. Note radial striations in the calvarium. B: Radiograph of the hand of the same patient. Note widened marrow cavities, thinned cortices, and osteoporosis. |
The skeletal dysplasia of thalassemia is related to iron chelation treatment. Iron chelation with desferrioxamine or oral deferasirox to prevent iron overload has dramatically impacted the health status of patients who require transfusions for thalassemia major (99, 100 and 101). Desferrioxamine, although essential in prolonging survival among transfusion-dependent patients, causes significant skeletal dysplasia in approximately 50% of cases (102). The findings include a slowing of spinal growth, biconcave vertebrae that progress to platyspondyly in some cases, and physeal widening at the wrist and knee that, in some patients, were severe enough to resemble rickets. Biopsies from patients with desferrioxamine-induced dysplasia show reduced and irregular bone mineralization as well as significant alterations in cartilage histology (103, 104). The spinal deformities are typically progressive, but metaphyseal lesions may heal with reduction of the desferrioxamine dose or following a switch to other iron chelators (105, 106). Skeletal dysplasias have not been reported with the newer oral iron chelator deferasirox (99, 107).
Osteopenia is a major skeletal manifestation of thalassemia major, occurring in more than 90% of patients despite optimal transfusion and chelation (108). Bone mineral density is lower in patients who have delayed puberty or amenorrhea (109, 110), indicating a possible role for endocrinopathy in the pathogenesis of osteopenia. Decreased bone density in patients with thalassemia is predominantly trabecular and associated with iron deposition (111). Consistent biochemical alterations in bone turnover have not been found (112). In patients with impaired sexual maturation, bone mineral density increases in response to hormone replacement therapy (113). In GH-deficient patients, GH administration can normalize markers of bone turnover but does not increase bone density (114). Bisphosphonates were ineffective in increasing bone mineral density in two placebo-controlled trials (115, 116).
Fractures are common in patients with thalassemia major, although they occur less often since the widespread use of young-onset transfusions began. The 40% to 50% incidence of fractures reported in some early series (117, 118 and 119) has decreased to 13% to 21% in recent series (120, 121 and 122). However, a multicenter review (121) found fractures to be often multiple or recurrent. The orthopaedist treating a fracture in a child with thalassemia should consider the problems of multiple fractures, weakened bone, high refracture risk, and clinically significant anemia.
The problems associated with transfusions and chelation have led to a search for alternative medical treatments for the thalassemias. Hydroxyurea, which stimulates hemoglobin F production, may prove effective (123), although at the time of writing this chapter, it has not been approved by the U.S. FDA for children with thalassemia. Bone marrow transplantation
has been used successfully in several centers for the treatment of severe thalassemia (124, 125 and 126), but it has not been shown to prevent osteopenia (109). Stem cell transplantation from umbilical cord blood of related donors has also been used with some success (127). Despite success in a mouse model (128), gene therapy for thalassemia is not yet a clinical reality.
has been used successfully in several centers for the treatment of severe thalassemia (124, 125 and 126), but it has not been shown to prevent osteopenia (109). Stem cell transplantation from umbilical cord blood of related donors has also been used with some success (127). Despite success in a mouse model (128), gene therapy for thalassemia is not yet a clinical reality.
DISORDERS OF NEUTROPHILS AND LYMPHOCYTES
The major cellular components of the immune system include neutrophils, lymphocytes, monocytes, and macrophages. Neutrophils serve as a first line of defense against bacterial and fungal diseases. Neutrophils circulate in the peripheral blood and, through a complex chemotactic process, migrate to sites of infection where they recognize, phagocytose, and kill pathogenic microorganisms. Lymphocytes are classified as B cells derived from bone marrow and T cells derived from the thymus. B cells control humoral immunity and T cells control cellular immunity. The complex interaction of the cells of the immune system is mediated largely through cytokines, and a discussion of this process is beyond the scope of this chapter. This section will discuss disorders of neutrophils [chronic granulomatous disease (CGD)], B cells [X-linked agammaglobulinemia (XLA)], and T cells (acquired immunodeficiency syndrome) that are relevant to the pediatric orthopaedist.
Chronic Granulomatous Disease.
CGD commonly causes recurrent, deep, bacterial or fungal infections of the musculoskeletal system, including osteomyelitis. Therefore, despite being a rare disease, CGD should enter the orthopaedist’s mind in the setting of atypical, unusually severe, or difficult-to-treat infections.
CGD is the most common congenital disorder affecting neutrophils, and occurs in approximately 1 in 200,000 to 1 in 500,000 live births (129). The disorder can be inherited in an X-linked recessive or autosomal recessive fashion. A key component of neutrophil function is the respiratory burst. After phagocytosis, creation of hydrogen peroxide and hypochlorous acid in the phagosome allows optimal killing of ingested pathogenic microorganisms. The creation of these oxidants is dependent on nicotinamide adenine dinucleotide phosphate (NADPH) oxidase. CGD is a group of disorders characterized by a variety of mutations of any of the NADPH component genes, transmitted in X-linked or autosomal patterns (130). The mean age at diagnosis depends on the type of CGD. X-linked CGD presents at a mean age of 3 years, whereas the autosomal recessive types typically present at 7 to 8 years (129). The diagnosis of CGD is made by detecting in vitro dysfunction of the respiratory burst (130).
The respiratory burst is particularly important in the killing of catalase-positive microorganisms. Catalase-positive organisms commonly encountered in infections in patients with CGD include S. aureus, gram-negative enteric bacteria, Burkholderia spp., Nocardia spp., and Aspergillus spp. (129). Infections with catalase-negative organisms such as S. pneumoniae and Haemophilus influenzae are uncommon because such organisms produce hydrogen peroxide that can be used by the neutrophil for killing when NADPH oxidase is ineffective.
Children with CGD typically present with recurrent bacterial and fungal infections (131). These infections can be superficial, such as lymphadenitis, infectious dermatitis, and perirectal abscesses, or deep, such as pneumonia, liver abscesses, and osteomyelitis. Also, the lack of the oxidant products of the respiratory burst, which would have acted to mediate or suppress further neutrophil chemotaxis, allows continued recruitment of neutrophils and the formation of granulomata.
Osteomyelitis occurs in approximately 25% of patients with CGD, according to a review of 368 patients in a national registry (129). Common sites include the hands and feet (132), as well as the spine and ribs (133). Aspergillus infections of the spine are typically difficult to treat. Some experts recommend surgical debridement with the wound left open to heal secondarily (133), although others have reported successful medical cure of spinal Aspergillus osteomyelitis using interferon-γ and antifungals (134, 135 and 136). One child with tibial Aspergillus osteomyelitis was successfully treated with interferon-γ and antifungals after failure of surgical debridement (137).
The treatment of established infections should be aggressive, with early surgical debridement and liberal use of antibiotics. Accurate cultures are essential, because pathogens uncommonly encountered in the general pediatric population are common causes of infection in patients with CGD. The possibility of fungal infection should always be specifically investigated with fungal smears and cultures.
Prophylaxis against infection is important in the treatment of children with CGD. Routine preventative measures such as hand-washing and good hygiene assume paramount importance in these individuals. Prophylactic administration of interferon-γ (138), itraconazole (139), or Bactrim helps to prevent infections.
Until recently, fewer than 50% of patients lived beyond the second decade of life (129); however, the long-term prognosis for children with CGD seems to be improving (140). Stem cell transplantation has been used with some success to “cure” CGD (141). Gene therapy has shown promise, but has yet to show lasting clinical efficacy (142). Bone marrow transplantation may soon have a role as well (143).
X-Linked Agammaglobulinemia.
XLA, an inherited defect in B cell maturation and function, may present to the orthopaedist as a clinical picture of arthritis. Arthritis occurs in XLA for unclear reasons and may be an initial presenting symptom in children at an average age of 2 years (144). Fifteen of sixty-nine patients in one series had arthritis at initial presentation; only four of these cases were due to infection (145). The arthritis of XLA most often affects the knees, wrists, ankles, and fingers, and may be polyarticular in presentation (146). The clinical picture may closely resemble juvenile rheumatoid arthritis (147). Synovitis is present, and the synovial tissue has a large number of suppressor
T lymphocytes, differentiating it pathologically from that of juvenile rheumatoid arthritis (148). Septic arthritis must be ruled out in both acute and chronic presentations because mycoplasmal infection was the leading cause of chronic arthritis in a series of 358 patients with XLA (149). The arthritis, if aseptic, usually responds to immune globulin treatment and anti-inflammatory medication (146). A knowledge of the clinical picture and consequences of XLA will allow the orthopaedist to appropriately refer patients for further evaluation and treatment.
T lymphocytes, differentiating it pathologically from that of juvenile rheumatoid arthritis (148). Septic arthritis must be ruled out in both acute and chronic presentations because mycoplasmal infection was the leading cause of chronic arthritis in a series of 358 patients with XLA (149). The arthritis, if aseptic, usually responds to immune globulin treatment and anti-inflammatory medication (146). A knowledge of the clinical picture and consequences of XLA will allow the orthopaedist to appropriately refer patients for further evaluation and treatment.
Initially reported by Bruton in 1953 as the first recognized primary immunodeficiency, XLA is otherwise known as Bruton agammaglobulinemia (150). XLA results from one of more than 750 possible mutations in the gene for B-lymphocyte tyrosine kinase, which is necessary for B cell maturation (151). The immunologic abnormality of XLA therefore consists of very low numbers of mature B cells and profoundly decreased production of all three major immunoglobulin classes (152). The number and function of T cells are usually normal.
Individuals with XLA are normal at birth, but as maternal IgG levels begin to decline in the first few months, recurrent infections begin to appear (153). Respiratory infections are common and are typically caused by organisms such as Streptococcus spp. and H. influenzae (144). Infections are usually severe enough to require hospitalization before a diagnosis of XLA is made (154). Therefore, the orthopaedist who is evaluating a child with unexplained arthritis should inquire about past history of hospitalization for respiratory or other infections. Infectious disease consultation should be obtained if an infection history accompanies a clinical picture of arthritis in young children.
The diagnosis of XLA is presumed in the setting of hypogammaglobulinemia and very low numbers of circulating B-lymphocytes. Treatment of XLA consists of immune globulin replacement and aggressive treatment of infections. Immune globulin, given as a regular prophylaxis, can lower the incidence of respiratory infections or other systemic infection and thereby prolong life (155). Recurrent respiratory infections lead to chronic lung disease, and respiratory failure is a major cause of mortality (153).
DISORDERS OF THE MONOCYTE-MACROPHAGE SYSTEM
The monocyte-macrophage system is a group of cell types derived from a common bone marrow precursor that provides important immune functions in various parts of the body. Macrophages ingest cellular debris, pathogens, and foreign bodies, and are particularly abundant in the spleen, liver, lymph nodes, lungs, and bone. Osteoclasts are a specialized form of macrophage, derived from the same precursor. Dendritic cells are nonphagocytic antigen-presenting cells that are thought to arise from the monocyte-macrophage stem cell. A wide variety of diseases affect the monocyte-macrophage system. Two diseases with musculoskeletal manifestations discussed in this chapter are Gaucher disease, which is a lysosomal storage disease, and Langerhans cell histiocytosis (LCH), which is a dendritic cell proliferative disorder.
Gaucher Disease.
Lysosomal storage disorders involve deficiencies of catabolic enzymes that allow toxic accumulation of metabolic pathway products. A variety of enzyme deficiencies lead to a variety of diseases with different manifestations. The most common lysosomal storage disease is Gaucher disease, and this example will be discussed in detail in this chapter. Gaucher disease has significant skeletal manifestations, and can require orthopaedic attention for bone pain, osteomyelitis, osteopenia, pathologic fractures, and ON.
In his doctoral thesis in 1882, Phillipe Charles Ernest Gaucher described a disease that causes splenic enlargement (156). The cause of the disease was not identified until 1965, when Brady et al. (157) linked it to a deficiency of glucocer-ebrosidase, a membrane-bound enzyme responsible for cleaving glucocerebroside. The lipid glucocerebroside accumulates in macrophages, and such lipid-laden macrophages are termed Gaucher cells. The clinical manifestations of Gaucher disease are caused by the accumulation of these cells in organs, resulting in organ dysfunction.
Gaucher disease is the most common inherited lysosomal storage disease, with an autosomal recessive inheritance pattern and a prevalence of 1 in 40,000 in the general population and 1 in 400 to 1 in 600 among Ashkenazi Jews (158, 159, 160 and 161). Three forms of Gaucher disease are recognized: type 1 (nonneuronopathic), type 2 (acute neuronopathic), and type 3 (subacute neuronopathic). Type 1 is by far the most common form and is characterized by hepatosplenomegaly, pancytopenia, and predominant skeletal manifestations. Type 2 is a rare form that involves the central nervous system and cranial nerves and usually causes death by apnea or aspiration before the age of 2 years (162). Type 3 disease is characterized by neurologic symptoms, including seizures, that begin during adolescence (163). More than 100 disease-producing mutations of the glucocerebrosidase gene, which is located on the short arm of chromosome 1, have been identified (163), and some mutations predict the phenotype (164). The detection of glucocerebroside in blood and urine confirms the diagnosis of Gaucher disease.
The age at onset and clinical presentation depend upon the genotype and clinical type. In a series of 53 patients, Zimran et al. (165) found that the average age at diagnosis was 25 years (range, 8 months to 70 years). Another series (164) of 34 children and adolescents with type 1 disease found that most of them presented before the age of 10 years. A patient with Gaucher disease may present initially to the orthopaedist with musculoskeletal symptoms. Bone pain or fracture is the reason for presentation in 13% to 60% of patients (165, 166). Growth retardation is also a common musculoskeletal presenting symptom, with 26% and 30% of patients presenting with less than the third percentile of normal values for weight and height, respectively (164). Skeletal abnormalities are detected radiographically in 88% to 94% of patients at presentation (164, 166).
The clinical manifestations of Gaucher disease depend on which organs are affected by accumulated Gaucher cells. Splenic
involvement causes splenomegaly and can cause hypersplenism, leading to anemia, thrombocytopenia, or pancytopenia. Liver involvement can cause mild liver dysfunction. Impaired hepatic synthesis of clotting factors may compound the thrombocytopenia, causing clinically significant coagulopathy.
involvement causes splenomegaly and can cause hypersplenism, leading to anemia, thrombocytopenia, or pancytopenia. Liver involvement can cause mild liver dysfunction. Impaired hepatic synthesis of clotting factors may compound the thrombocytopenia, causing clinically significant coagulopathy.
Skeletal involvement is a prominent feature in Gaucher disease and substantially impacts the quality of life (165, 167). In a review of 602 patients with type 1 Gaucher disease from the Gaucher registry, 21% were found to have some form of disability in mobility related to skeletal involvement (168). Skeletal manifestations include pain, deformity, osteopenia, ON, osteomyelitis, pathologic fracture, and vertebral collapse (167). Gaucher disease is associated with a classic abnormality that shows up on radiographs as an “Erlenmeyer flask” deformity of the distal femur and proximal tibia, representing impairment of remodeling (Fig. 10-5). However, this finding is not pathognomonic for Gaucher disease and occurs only in 56% to 70% of patients with known Gaucher disease (165, 166).
Bone crises are a common symptom of skeletal involvement. Bone crises are thought to be related to intramedullary or subperiosteal hemorrhage (169, 170) made possible by thrombocytopenia and deficient clotting factor synthesis. Bone crises are episodes of acute bone pain accompanied by fever, leukocytosis, and elevated erythrocyte sedimentation rate. Because of this clinical picture, bone crises are also known as pseudoosteomyelitis. Blood cultures may help differentiate between bone crisis and osteomyelitis. Further differentiation is difficult, as in SCD, and imaging studies may not be helpful. Early in bone crises, plain radiographs are normal, but may progress to show periosteal reaction and areas of radiolucency (171, 172). Radionuclide bone scans may show an area of decreased uptake early in the course of the process (173) and increased uptake around a photopenic area later in the course (174). MRI shows marrow edema on T2-weighted images, with or without signs of hemorrhage (168, 170). Periosteal fluid accumulation seen on MRI may indicate infection and should be aspirated for culture under sterile conditions and radiographic guidance.
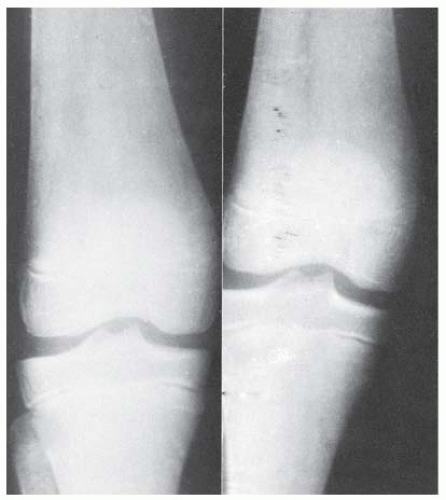 FIGURE 10-5. Radiographs of the knee in a child with Gaucher disease. Note the typical flaring of the distal femoral metaphysis, or Erlenmeyer flask deformity. (Photo courtesy of Henry J. Mankin, MD.) |
Treatment of bone crises is supportive. Severe pain early in the course usually requires opioid analgesics, which can be augmented with high-dose prednisolone (175). The symptoms gradually abate over 2 to 4 weeks. Failure of the symptoms to improve should warrant further investigation into the possibility of osteomyelitis. Bone aspiration in an operating room setting may be required. Osteomyelitis can follow a bone crisis, often with anaerobic organisms, suggesting that there has been a period of ischemia (176). Treatment of osteomyelitis in Gaucher disease parallels that in SCD as discussed earlier, although attention should be paid to the altered structural integrity of bone and increased bleeding risk when surgical debridement is considered in a patient with Gaucher disease. ON can follow a bone crisis, so routine radiographic evaluation of an affected area is necessary even after the crisis resolves.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


